48uep6bbphidvals|399
48uep6bbph|2000F98CTab_Articles|Fulltext
Introduction
Gastrointestinal stromal tumours (GIST) are mesenchymal tumours of the gastrointestinal tract, accounting for approximately 0.2-1%[1,2] of gastric malignancies. Primary GIST can originate from all regions of the GI tract most commonly being found in the stomach (40-70%), small bowel (20-40%), colorectum (5-15%) and esophagus (5%)[11]. We report one such case of a large malignant irresectable stromal tumour of the stomach, feigning as pancreatic pseudocyst, associated with the presence of concomitant distant metastases, treated palliatively, and followed by adjuvant imatinib therapy.
Case Report
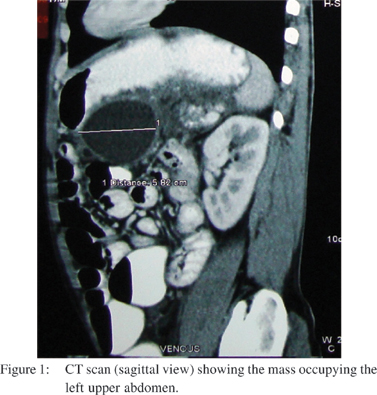
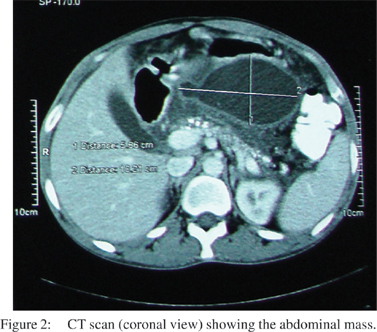
A 48 year old male, an alcoholic, presented to the surgery department of our hospital with vague abdominal pain and a large lump occupying the left side of the abdomen. Clinically, a large lobulated lesion was palpable in the upper abdomen, about 15x10x8 cm, predominantly involving the left upper half, and appearing to extend up to the pelvic inlet. Ultrasonography showed a heterogeneous mass size of 16x14 x12 cm, with cystic necrotic components interspersed. A pseudocyst of the pancreas was taken as a possible pre-operative differential diagnosis. A CT scan confirmed the presence of a 10x6 cm mass occupying the left upper abdomen (Figures 1,2). Upper GI endoscopy revealed a large globular ulcerated lesion with central umblication at the posterior wall of the body of the stomach. A USG guided biopsy was done and a gastrointestinal stromal tumour was reported.
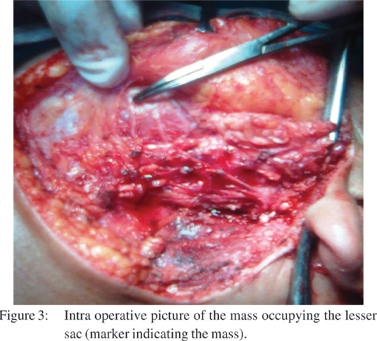
An exploratory laprotomy was done, and intraoperatively, a large cystic lesion of size 15x10 cm, occupying the lesser sac, densly adherent to the posterior wall of the stomach and transverse colon and extending downward up to pelvic inlet was present (Figure 3). In addition, perigastric lymph nodes were also found to be involved. Intra-tumoral evacuation of haemorrhagic, necrotic material with intra-lesional drain placement in the lesser sac, as well as lymph node biopsy was done, and specimens were sent for histopathology.
Microscopy showed elongated spindloid cells with hyperchromatic nuclei and areas with a whorled pattern, while the cyst wall showed fibro-fatty tissue with smooth muscle bundles infiltrated with dense round cells. Few spindloid cells with hyperchromatic nuclei were seen, suggestive of a gastrointestinal stromal tumour of the stomach. Postoperative period was uneventful, and patient was commenced on imatinab mesylate.
Discussion
Gastrointestinal stromal tumors are the most common sarcomas of the gastrointestinal tract, accounting for 0.2% of all GI tumours.[1,2] The term gastrointestinal stromal tumor (GIST) was first used by Mazur and Clarke in 1983, to describe the nonepithilial tumours of the GI tract that lack the ultrastructural feature of smooth muscle cells as well as the immunohistochemical characteristics of Schwann cells.[3] Based on their histologic and immunohistochemical features, GIST are presumed to originate from the interstitial cells of Cajal (ICC), which are components of the intestinal autonomic nervous system and act as pacemakers, regulating intestinal peristalsis.[4]
In 1998, Hirota et al demonstrated the involvement of a functional mutation of the Kit proto-oncogene.[5] Kit is a tyrosine kinase receptor that gets activated when bound to a ligand known as ‘steel factor’.[6] Oncogenic mutations of Kit have been found in neoplasms associated with mast cell tumours, myelofibrosis, germ cell tumours, GIST, etc.[7] GIST are identified by the universal expression of the CD117 antigen (95%).[7] GIST have been found to be associated with functional mutations in the PDGFRA tyrosine kinase receptor.[8] In addition, transition from ICC hyperplasia to low risk GIST is associated with 14q deletion; while loss of 22q is found in high risk or metastatic GIST.[9] The broad morphological spectrum exhibited by GIST at a light microscopic and ultrastructural level has generated much debate and controversy concerning tumour histogenesis. Although this will probably remain for the foreseeable future, Kindblom et al[10] and more recently Sircar et al[11] have elegantly provided considerable support for a possible histogenetic origin from the ICCs encountered in the gastrointestinal tract, which are thought to play a role in coordinating intestinal motility.[12,13]
The ICCs appear to be modified smooth muscle cells occurring at various intramural sites within the intestinal tract, primarily in the muscularis propria and in association with the myenteric plexus.
Sites outside gastrointestinal tract that express CD 117/ckit are melanocytes, basal cells of epidermis, immature Langerhans cells in the epidermis, a variety of epithelial cells (breast/salivary gland/sweat gland/renal tubule), cells present in the reproductive system, a subset of glial cells, and osteoclast precursors. Although CD117 is a diagnostically useful antigen expressed by the interstitial cells of Cajal (and in most gastrointestinal stromal tumours), it is important to be aware of the expression of this antigen in a variety of other cells.
Primary GIST can originate from all regions of the GI tract but are most commonly found in the stomach (40-70%), small intestine(20-40%), colorectum (5-15%) and esophagus (5%).[14] They are equally common in men and women and usually present in the 40-60 age group.[15] Association of paragangliomas, pulmonary chondromas, and gastric lesions, called as ‘Carney’s triad’, was initially classified as leiomyosarcoma.[16] Many GIST are asymptomatic and discovered incidentally during imaging and laparotomy for other reasons. In advanced diseases, the patient may present with a mass lesion or with vague abdominal discomfort, although the tumour can grow to a very large size before producing any symptoms. GIST can be highly vascular and bleeding is one of the most common symptoms. These
tumours are typically soft and friable and can cause life threatening haemorrhage.
Tumour rupture along with intraperitoneal bleeding can occur, leading to a high risk of dissemination by peritoneal seeding of the tumour. Obstruction of the GI tract is occasionallya presenting condition and can lead to perforation. GIST with overt metastatic diseases, account for 5-15% of all cases.[14,17] The most common metastatic sites are the liver and peritoneum. Diffuse peritoneal spread is not uncommon and may involve innumerable small tumour nodules essentially replacing the omentum, studding the diaphragmatic surface or covering the serosal surface of the bowel.
GIST, gastrointestinal leiomyoma or leiomyosarcoma, schwannoma, local extension by a primary retroperitoneal dedifferentiated liposarcoma, benign and malignant vascular tumours, intra-abdominal fibromatosis (desmoid tumour), carcinoid with a spindle cell morphology, and metastatic disease (spindle cell melanoma/spindle cell carcinoma) are the predominant tumours that may need to be considered in the differential diagnosis. Most of these tumours can be characterized accurately on the basis of precise clinical data and diligent microscopy, supplemented by appropriate immunohistochemical, ultrastructural, and molecular biological analyses. Separating GIST from true smooth muscle tumours is clinically relevant because differences in biological behavior can sometimes pose difficulties in differentiation.
These lesions cannot be easily differentiated from other GI tumours of smooth muscle origin. However, because of their submucosal location a fine–needle aspiration or core biopsy with endoscopic ultrasound guidance is commonly required to obtain tissue for diagnosis.[18] CT scans are used to determine the anatomic extent of the GIST and to assist with operative planning. Unfortunately CT is unable to differentiate between inflammatory adhesions and malignant involvement of an adjoining organ, and is unlikely to identify any peritoneal metastasis smaller than 2 cm in diameter. PET scan is useful along with CT for evaluating GIST in totality as well as for assessing response to chemotherapy. Sequential scans prior to and after imatinib mesylate therapy, reliably and rapidly indicate the responsiveness or resistance of the tumour to the drug.[19]
GIST exhibit a wide spectrum of clinical behaviour and may remain stable for years together. Primary tumours more than 5 cm in size and those with increased mitotic activity are associated with poor prognosis. Location of the tumour in the small bowel further worsens the outcome.[1,20]
Surgery remains the gold standard therapy for all resectable tumours. The importance of clear resection of margins cannot be emphasized. Tumour rupture or any trauma to the pseudocapsule is associated with an enormously increased risk of recurrence, including a risk of dissemination of tumour throughout the peritoneum. If lesions involve adjacent organs, en-bloc resection must be done in order to avoid further spillage. Overall 5 year survival rates after complete resection of localized GIST are 40-55%.[21,22,23,24] Lymph node metastases are extremely rare so that regional lymphadenectomy is not recommended routinely. Lymph node dissection should be undertaken for patients with any evidence or suspicion of nodal metastasis. The typical site of tumour recurrence following resection of GIST is the liver and peritoneum. Pulmonary metastases are rare. Mostly abdominal/pelvic CT scans are used for post treatment follow up.
Although it is not well documented which group of patients will benefit most from imatinib treatment, or the appropriate duration of treatment, its success as an orally administered inhibitor of the Kit receptor tyrosine kinase in managing metastatic disease has lead to an improvement of resectability, especially in cases of technically inoperable primary tumours. Responses to imatinib are evident in 30-40% of patients after 7 months and 50-60% after 9 months of treatment.[25] Surgery is performed when tumour size has decreased to the point at which resection is technically possible or when successive CT scans show no further evidence of tumour shrinkage.
References
1. Fletcher CD, Berman JJ, Corless C, Gorstein F, Lasota J, Longley BJ, et al. Diagnosis of gastrointestinal stromal tumors: A consensus approach. Hum Pathol. 2002;33:459–65.
2. Jemal A, Murray T, Ward E, Samuels A, Tiwari RC, Ghafoor A, et al. Cancer statistics, 2005. CA Cancer J Clin. 2005;55:10–30.
3. Mazur MT, Clark HB. Gastric stromal tumors. Reappraisal of histogenesis. Am J Surg Pathol. 1983;7:507–19.
4. Kindblom LG, Remotti HE, Aldenborg F, Meis-Kindblom JM. Gastrointestinal pacemaker cell tumor (GIPACT): gastrointestinal stromal tumors show phenotypic characteristics of the interstitial cells of Cajal. Am J Pathol. 1998;152:1259–69.
5. Hirota S, Isozaki K, Moriyama Y, Hashimoto K, Nishida T, Ishiguro S, et al. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science. 1998;279:577–80.
6. Fleischman RA. From white spots to stem cells: the role of the Kit receptor in mammalian development. Trends Genet. 1993;9:285–90.
7. Rubin BP, Singer S, Tsao C, Duensing A, Lux ML, Ruiz R, et al. KIT activation is a ubiquitous feature of gastrointestinal stromal tumors. Cancer Res. 2001;61:8118–21.
8. Heinrich MC, Corless CL, Duensing A, McGreevey L, Chen CJ, Joseph N, et al. PDGFRA activating mutations in gastrointestinal stromal tumors. Science. 2003;299:708–10.
9. Corless CL, Fletcher JA, Heinrich MC. Biology of gastrointestinal stromal tumors. J Clin Oncol. 2004;22:3813–25.
10. Kindblom LG, Remotti HE, Aldenborg F, Meis-Kindblom JM. Gastrointestinal pacemaker cell tumor (GIPACT): gastrointestinal stromal tumors show phenotypic characteristics of the interstitial cells of Cajal. Am J Pathol. 1998;152:1259–69.
11. Sircar K, Hewlett BR, Huizinga JD, Chorneyko K, Berezin I, Riddell RH. Interstitial cells of Cajal as precursors of gastrointestinal stromal tumors. Am J Surg Pathol. 1999;23:377–89.
12. Thuneberg L. Interstitial cells of Cajal: intestinal pacemaker cells? Adv Anat Embryol Cell Biol. 1982;71:1–130.
13. Hagger R, Finlayson C, Jeffrey I, Kumar D. Role of the interstitial cells of Cajal in the control of gut motility. Br J Surg. 1997;84:445–50.
14. Bauer S, Corless CL, Heinrich MC, Dirsch O, Antoch G, Kanja J, et al. Response to imatinib mesylate of a gastrointestinal stromal tumor with very low expression of KIT. Cancer Chemother Pharmacol. 2003;51:261–5.
15. Chompret A, Kannengiesser C, Barrois M, Terrier P, Dahan P, Tursz T, et al. PDGFRA germline mutation in a family with multiple cases of gastrointestinal stromal tumor. Gastroenterology. 2004;126:318–21.
16. Carney JA. Gastric stromal sarcoma, pulmonary chondroma, and extra-adrenal paraganglioma (Carney Triad): natural history, adrenocortical component, and possible familial occurrence. Mayo Clin Proc. 1999;74:543–52.
17. Roberts PJ, Eisenberg B. Clinical presentation of gastrointestinal stromal tumors and treatment of operable disease. Eur J Cancer. 2002;38 Suppl 5:S37–8.
18. Rader AE, Avery A, Wait CL, McGreevey LS, Faigel D, Heinrich MC. Fine-needle aspiration biopsy diagnosis of gastrointestinal stromal tumors using morphology, immunocytochemistry, and mutational analysis of c-kit. Cancer. 2001;93:269–75.
19. Stroobants S, Goeminne J, Seegers M, Dimitrijevic S, Dupont P, Nuyts J, et al. 18FDG-Positron emission tomography for the early prediction of response in advanced soft tissue sarcoma treated with imatinib mesylate (Glivec). Eur J Cancer. 2003;39:2012–20.
20. Miettinen M, El-Rifai W, L HLS, Lasota J. Evaluation of malignancy and prognosis of gastrointestinal stromal tumors: a review. Hum Pathol. 2002;33:478–83.
21. DeMatteo RP, Lewis JJ, Leung D, Mudan SS, Woodruff JM, Brennan MF. Two hundred gastrointestinal stromal tumors: recurrence patterns and prognostic factors for survival. Ann Surg. 2000;231:51–8.
22. Joensuu H, Fletcher C, Dimitrijevic S, Silberman S, Roberts P, Demetri G. Management of malignant gastrointestinal stromal tumours. Lancet Oncol. 2002;3:655–64.
23. Crosby JA, Catton CN, Davis A, Couture J, O’Sullivan B, Kandel R, et al. Malignant gastrointestinal stromal tumors of the small intestine: a review of 50 cases from a prospective database. Ann Surg Oncol. 2001;8:50–9.
24. Pierie JP, Choudry U, Muzikansky A, Yeap BY, Souba WW, Ott MJ. The effect of surgery and grade on outcome of gastrointestinal stromal tumors. Arch Surg. 2001;136:383–9.
25. Demetri GD, von Mehren M, Blanke CD, Van den Abbeele AD, Eisenberg B, Roberts PJ, et al. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med. 2002;347:472–80.