48uep6bbphidvals|183
48uep6bbphidcol2|ID
48uep6bbph|2000F98CTab_Articles|Fulltext
Deciphering the 3 billion nucleotides long human genome through direct sequencing that culminated in 2001, in fact ahead of schedule, was a jewel in the crown of human geneticists. [1] And the hallowed yet seemingly imponderable precincts of genetics were conquered but only to further incite the insatiable human curiosity. The marvellous insight into the hidden truths of nature revealed that there are about 25,000 genes in the human genome. As we know these genes are embedded long stretches of DNA which rearrange into a pair of 23 chromosomes during the cell cycle for even distribution of the genome to daughter cells. These genes are interspersed within a large non-coding DNA sometimes referred to as ‘junk’ DNA (more out of ignorance than otherwise). The human race is relatively of recent origin being the last during the 3.5 billion or more of evolution of life on earth attesting to the complexity or the superiority of being a human (at least humans tend to believe that!). Evolutionary changes in the human genome are relatively rare events to the tune of one nucleotide change in 10[8] nucleotides per generation. [2] Thus, there are remarkable similarities in the human genome across different populations despite obvious phenotypic differences. There are however, recombination events in the genome during the meiosis between different segments of 2 chromosomes so that the offspring inherits a mixture of information from both the paternal and maternal genes. For distantly located genes on different chromosomes, the frequency of a genotype will depend on the frequencies of alleles (alternate forms of a gene; Figure 1) in a large random mating population. For example, for 2 different genes (e.g. A and B) having 2 alleles each (A1, A2 and B1, B2), the chance of an individual possessing a particular genotype (A1B1, A1B2, A2B1, A2B2) depends on the frequency of the alleles in the population e.g. for frequencies of A1 90%, A2 10%, B1 95%, B2 5%, the chances of genotypes A1B1, A1B2, A2B1, A2B2 will be 85.5%, 4.5%, 9.5%, and 5% respectively. This is known as linkage equilibrium where the frequencies of genotype at one locus are independent of the frequencies at another locus. However, closer loci on one chromosome are in what is known as linkage disequilibrium (LD).[3] Linkage disequilibrium is a result of recent mutations or polymorphisms in that particular area that have not been subjected to meiotic recombination events over a long period of time and thus the chances of 2 neighbouring alleles inherited together are more than that in linkage equilibrium. The recombination events occur at certain spots separated by relatively long distance in the chromosome. Thus, the part of the chromosome in between the recombination sites is passed on to the next generation en bloc. The genomic elements e.g. genes or short tandem repeats (microsatellites) within such a block are linked physically and are in LD. Linkage disequilibrium is the basis of genetic association studies.

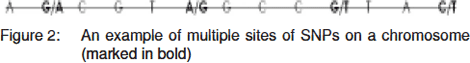
Although human genome is similar between individuals to a great extent, we know that there are dissimilarities due to occurrence of single base pair changes at multiple locations across the genome as part of the evolutionary changes often conferring a survival advantage. These single base pair changes are popularly known as single nucleotide polymorphisms or SNPs (Snips) (Figure 2). The SNPs are located almost at regular intervals ~one after every 300 base pairs with an estimated 10 million SNPs across 3 billion nucleotides in the human genome with both alleles having a frequency of at least 1%.[4] Different alleles of a gene are often a result of SNPs and most genes are biallelic. The SNPs arise on an existing background
over a period of time and are in LD with each other over a short stretch of a chromosome and are thus co-inherited. A collateral benefit of greater significance of the human genome project was the International Human Hapmap Project whose 1st phase was completed in 2005 and the 2nd phase in 2007.[5,6] The data are in public domain freely available to every one. A haplotype is a combination of alleles or a pattern of SNPs at multiple sites on a single chromosome, all of which are transmitted together because they are in linkage disequilibrium with each other (Figure 3). The hapmap (haplotype map) was designed to determine the frequencies and patterns of association among roughly 3 million common SNPs. The human hapmap was created by genotyping 1 million SNPs in phase 1 and 3 million SNPs in phase 2 in samples of 270 individuals from 4 different ancestry- 30 trios (mother, father and child) from Yoruba in Ibadan, Nigeria; 30 trios from Utah residents of Northern and Western European origin; 45 unrelated Chinese and 45 unrelated Japanese. Once we know the orderly arrangement of SNPs on a chromosome, we can easily understand that if we can find out the presence of some of the SNPs on a chromosome, we can predict the presence of other SNPs in the vicinity which are in linkage disequilibrium with those SNPs. Some of these SNPs might be in the exon of a gene or gene regulatory sequence and may alter the function of the gene resulting in susceptibility for a disease. Because of the linkage disequilibrium, much fewer SNPs will give information about the nearby SNPs and other genomic areas e.g. genes of interest (risk alleles) conferring an increased risk of a particular disease. These SNPs serve as genomic markers because they reveal the presence of genes located near them in the haplotype block and are known as tag SNPs.[7] A haplotype block is ~11-22 kb bases long. Hapmap was designed to study common disease causing variants based on the ‘common disease-common variant hypothesis’ that means that the genetic susceptibility is due to allelic variants that have an allelic frequency of 1-5% in the population. [7]

Many human diseases have a genetic basis. Searching for a causal gene for a particular disease may prove to be very difficult. One general approach to search for a causal gene is through candidate gene analysis. This is based on a biologically plausible hypothesis of the likely mechanism for that disease and then searching for any mutations in the putative gene of interest. Genetic mutations in single gene disorders that follow Mendelian pattern of inheritance were discovered in this way e.g. mutations in genes for CFTR and haemophilia. However, such an approach is difficult to follow and unlikely to yield useful information in complex polygenic disorders with strong gene-gene and gene- environment interactions because a single hypothesis is unlikely to explain the disease process. In such a scenario, hypothesis independent approach to identify the genes involved in the pathogenesis of a disease is required. However, it will be difficult to search for such genes blindly unless one tries to sequence the entire genome in a large number of affected patients, an expensive and time consuming affair. An easier and feasible answer to this problem was provided by the study of variations in genetic markers. Microsatellite markers, which are 2 or 4 bases short tandem repeats (e.g. CACACACACA— CA) and are spaced across the genome after every 1-2 megabases, were used earlier in linkage analysis studies.[8] The problem with microsatellite markers is that it is a tedious approach because one has to study various microsatellite markers one by one. Moreover, even after finding a particular marker as being linked to the disease associated gene, it is difficult to find out or map the causal mutated gene in the nearby location that spans at least a few megabases (or ~10 cM). As opposed to the microsatellite markers, a study of SNPs as genetic markers through hapmap provides a much better and easier approach. Since SNPs are in linkage disequilibrium, it is estimated that 300,000 to 1 million well chosen tag SNPs should be able to provide information about most of the disease causing genes or gene regulatory sequences located closer to these SNPs.[7] By carefully selecting the number and locations of these tag SNPs based on their LD pattern, one can compare the SNPs between patients and controls to identify the differences in the frequencies of SNPs. For example, suppose a disease associated gene X is located somewhere on the short arm of chromosome 5 about which there is no a priori knowledge. A SNP that is located closer to and is in LD with the gene X is likely to be present in patients suffering from that disease significantly more commonly than in controls. After finding out such a SNP on the short arm of chromosome 5, one can then narrow down the search around that SNP over a relatively short distance of a few thousand bases to find out the mutation in gene X by direct sequencing of that region of the genome (known as resequencing) (Figure 4). To test for a large number of SNPs, chip based hybridization array technologies are used. Initially, chips containing 100,000 SNPs were used but currently chips with 1 million SNPs are available. Chips contain multiple short oligonucleotide probes having the sequence of the different SNP bearing regions. These many SNPs spaced across the length of the genome are supposed to cover the entire genome. If one studies the association of a particular disease with these many SNPs by comparing their frequencies in patients and controls, one is quite likely to find some significant associations between the disease and SNPs. This is the basis of genome wide association studies (GWAS).[9] There has been quite a lot of excitement about the possibility of finding the causal genes for various diseases such as diabetes, cardiovascular diseases, cerebrovascular accidents, Alzeihmer’s disease etc and quantitative traits such as height through GWAS.7 In fact, many studies have been published during a short period of the past 2-3 years and the readers of prestigious journals such as NEJM and Nature Genetics are inundated with at least one study detailing the GWA in every other issue. I will briefly mention here important GWA studies related to 3 common GI diseases:

Crohn’s disease: A number of GWA studies have been carried out to find out an association of SNPs with Crohn’s disease. Besides NOD2 and IBD5 locus on chromosome 5q31, many studies have shown strong association with 3 other genes namely IL23R, ATG16L1 and IRGM.[10,11,12] Interleukin 23 receptor is involved in mucosal inflammation. Both ATG16L1 and IRGM genes are related to autophagy and this association has revealed another important pathogenetic mechanism involved in Crohn’s disease. It is quite likely that the downstream pathways associated with these genes will soon lead to a better understanding of the disease pathogenesis and treatment.
Hepatitis B virus infection: A recent study from Japan in 786 Japanese chronic hepatitis B cases and 2,201 controls identified 11 SNPs in a region including HLA-DPA1 and HLADPB1.[13] Further validation study in Japanese and Thai patients, and controls showed that the susceptibility to chronic hepatitis B virus infection is associated with risk haplotypes (HLADPA1(*) 0202-DPB1(*)0501 and HLA-DPA1(*)0202-DPB1(*)0301, OR = 1.45 and 2.31, respectively) and protective haplotypes (HLA-DPA1(*)0103-DPB1(*)0402 and HLADPA1(*) 0103-DPB1(*)0401, OR = 0.52 and 0.57, respectively). Such studies help explain the genetic reasons why some individuals fail to clear the infection and develop chronic hepatitis B through disturbances in their immune mechanisms involved in virus clearance.
Primary biliary cirrhosis: In a recent GWAS analysis, 536 patients with primary biliary cirrhosis (PBC) and 1536 controls from Canada and US were genotyped for more than 300,000 SNPs.[14] Significant associations were shown between PBC and 13 loci across the HLA class II region; the HLA-DQB1 locus had the strongest association (P=1.78x10-19; odds ratio 1.75). Significant and reproducible associations were also found with two SNPs at the IL12A locus (encoding IL-12á) –rs6441286 (P = 2.42x10-14, odds ratio 1.54) and rs574808 (P=1.88x10-13, odds ratio 1.54), and one SNP (rs3790567) at the IL12RB2 locus (encoding interleukin-12 receptor â2) (P=2.76x10-11, odds ratio, 1.51). Fine-mapping analysis showed that a five-allele haplotype in the 3' flanking region of IL12A was significantly associated with PBC (P=1.15x10-34). The data suggested that the interleukin-12 immunoregulatory pathway might be important in the pathophysiology of PBC.
Problems with GWAS: GWAS is not the final answer and indeed there are many problems with GWAS analyses which include (i) the need for a large sample size. This is required because of 3 reasons: small effect size of the associated common SNPs, low minor allele frequency, and the large number of SNPs being tested,[8] (ii) the need for strict phenotypic characterization of patients and controls, (iii) the need for adjusting the p value for multiple comparisons e.g. with Bonferonni method.[8] The p value should be divided by the number of SNPs tested e.g. a p value of 0.05 should be divided by 100,000 if that many SNPs have been compared between patients and controls. Thus, a p value of 5X10-7 or 10-8 is considered significant. (iv) small effect size of the risk allele with an odds ratio of 1.2-1.5 in most studies suggesting only minor contribution of the risk allele to the causation of disease[15] (v) since the pattern of LD among SNPs is based on data from 4 populations, it is likely that the pattern might differ in another population with different ancestry e.g. Indian population. A recent study has indeed shown that there are important differences between SNPs in Indians and reported data in the Hapmap project.[16] Moreover, there are differences among different sub-populations in India. In the latest phase of hapmap project however, samples from many other populations including US residing Gujarati Indians have been included and the results should be available in near future (vi) another problem with the GWAS analysis is that most often the disease associated SNPs are found within a region where no known gene is present since 98% of the genome is noncoding and thus the functional significance of such an association is not well understood. However, with recent projects such as Encode (Encyclopedia of DNA Elements), we are beginning to find out more about such regions which contain important transcription sites and regulatory elements, and (vii) it is an expensive project. For example, with a sample size of 1000 patients and 1000 controls, the cost of chips and related consumables will be approximately $6-800,000 (INR 3-4 crores) even though the cost has come down substantially, and this does not include the hardware and manpower costs.
GWAS is only a first step: One needs to understand that through GWAS we can only find out an association with SNPs and usually not the causal gene. That is why some people call it a fishing expedition. There are many steps before a gene can be implicated in the disease pathogenesis after the GWAS analysis finds a significant association with SNPs. These include (i) confirming the association of the SNPs in a replication cohort of patients ideally from a different ethnic background, (ii) doing what is called fine mapping. Fine mapping is like combing the haplotype block where the SNP is located to find out the relevant gene or gene regulatory sequences having a mutation/polymorphism which might have a biologically plausible role in the pathogenesis of the disease in question, (iii) integrating the function of that gene in a relevant cellular pathway that might be involved in the pathogenesis of the disease, and finally (iv) performing functional studies in cell lines or experimental animals to show whether or not dysfunction of that particular gene results in similar phenotype (disease). Thus, one has to work backwards to nail the culprit.
What next? There is growing realisation that ultimately the best way to find out the risk associated gene is through whole genome sequencing. It is hoped that with the availability of 2nd generation sequencers, the cost for the whole genome sequencing will come down to about $1000 per sample by 2010 and then whole genome sequencing will be become more cost effective, and obviously a more direct and better way of finding the mutation(s) causing the disease including the rare alleles conferring high risk for the disease.
These are exciting times for understanding the genetic basis and surprising new biological pathways of complex diseases which have been long shrouded in mystery. In this bicentennial celebration year of Darwin’s birthday and his Theory of Evolution, genetics, though never out of vogue, is back in fashion. But why Darwin missed out, despite observing, on what Mendel mapped out later for fame is a gossip time fascinating tale.
References
1. International Human Genome Sequencing Consortium. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921.
2. International HapMap Consortium. The International HapMap Project. Nature. 2003;426:789–94.
3. Slatkin M. Linkage disequilibrium—understanding the evolutionary past and mapping the medical future. Nat Rev Genet. 2008;9:477–85.
4. www.ncbi.nlm.nih.gov/projects/SNP/
5. International HapMap Consortium. A haplotype map of the human genome. Nature. 2005;437:1299–320.
6. International HapMap Consortium. 2007. A second generation human haplotype map of over 3.1 million SNPs. Nature. 449:851–61.
7. Manolio TA, Brooks LD, Collins FS. A HapMap harvest of insights into the genetics of common disease. J Clin Invest. 2008;118:1590–605.
8. Lewis CM. Genetic association studies: design, analysis and interpretation. Brief Bioinform. 2002;3:146–53.
9. Hirschhorn JN, Daly MJ. Genome-wide association studies for common diseases and complex traits. Nat Rev Genet. 2005;6:95–108.
10. Duerr RH, Taylor KD, Brant SR, Rioux JD, Silverberg MS, Daly MJ, et al. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science. 2006;314:1461–3.
11. Rioux JD, Xavier RJ, Taylor KD, Silverberg MS, Goyette P, Huett A, et al. Genome-wide association study identifies new susceptibility loci for Crohn’s disease and implicates autophagy in disease pathogenesis. Nat Genet. 2007,39:596–604.
12. Parkes M, Barrett JC, Prescott NJ, Tremelling M, Anderson CA, Fisher SA, et al. Sequence variants in the autophagy gene IRGM and multiple other replicating loci contribute to Crohn’s disease susceptibility. Nat Genet. 2007;39:830–2.
13. Kamatani Y, Wattanapokayakit S, Ochi H, Kawaguchi T, Takahashi A, Hosono N, et al. A genome-wide association study identifies variants in the HLA-DP locus associated with chronic hepatitis B in Asians. Nat Genet. 2009;41:591–5.
14. Hirschfield GM, Liu X, Xu C, Lu Y, Xie G, Lu Y, et al. Primary Biliary Cirrhosis Associated with HLA, IL12A, and IL12RB2 Variants. N Engl J Med. 2009 May 20. [Epub ahead of print]
15. Goldstein DB. Common genetic variation and human traits. N Engl J Med. 2009;360:1696–8.
16. Indian Genome Variation Consortium. Genetic landscape of the people of India: a canvas for disease gene exploration. J Genet 2008;87:3–20.