48uep6bbphidvals|425
48uep6bbph|2000F98CTab_Articles|Fulltext
Introduction
MNGIE (mitochondrial neuro gastrointestinal encephalopathy) is a complex, autosomal-recessive, multisystem disorder with gastrointestinal dysmotility and severe cachexia as clinical hallmarks. Prior names given to this syndrome include polyneuropathy, ophthalmoplegia, leukoencephalopathy, and intestinal pseudo-obstruction (POLIP) syndrome; oculogastrointestinal muscular dystrophy (OGIMD); and mitochondrial encephalomyopathy with sensorimotorpolyneuropathy, ophthalmoplegia and pseudo-obstruction (MEPOP). The aim of this case presentation is to highlight the consideration of MNGIE in patients of intestinal pseudoobstruction who have concomitant involvement of the nervous system. Therapeutic options are limited, focusing on symptomatic control and adequate nutritional support.
Case Report
A 25 year old female presented with chief complaints of abdominal pain, vomiting and headache since 9 months. The abdominal pain was described as a dull ache predominantly in the upper abdomen and peri-umbilical region, and was present continuously. There was no radiation or postural variation of pain. The pain was not associated with any visible peristalsis. The pain used to be aggravated after meals and improved with vomiting. Vomiting was bilious, non projectile in nature. She also had early satiety and bloating of abdomen after meals. She also complained of amenorrhea since 6 months, decreased appetite and weight loss. On examination, the patient was cachectic with weight of 22 kg, height of 147 cm and BMI 12.24.
Abdominal examination was normal except for the sluggish bowel sounds. Neurological examination revealed partial ptosis of the left eye, bilateral lateral gaze palsy and bilateral grade I nystagmus. Rest of the systemic examination was normal. So the clinical picture of proximal small bowel obstruction with central nervous system involvement was suggested. The differential diagnosis included disseminated tuberculosis and lymphoma.
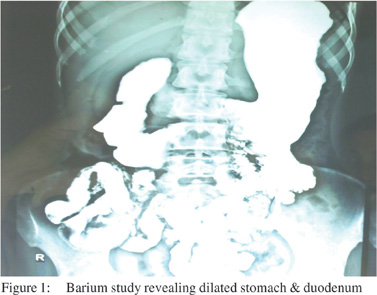
Investigations of the patient showed a normal hemogram and ESR was 5 mm/hr. Blood sugar and renal function tests were normal. Liver function tests were normal except a serum albumin of 2.5 g/dl. Thyroid profile was normal. HIV was negative. X ray abdomen showed dilated stomach and small bowel loops. Barium study revealed a dilated stomach and duodenum (Figure 1).
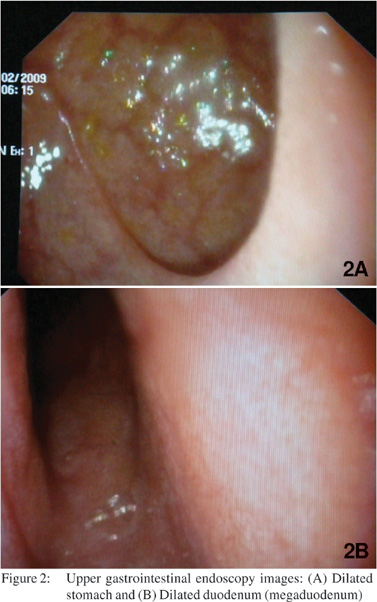
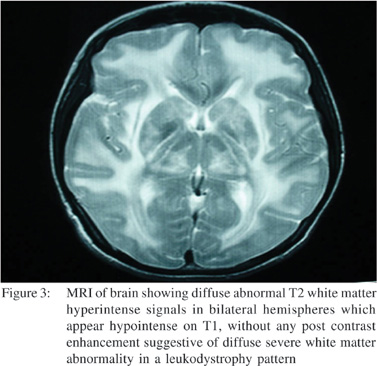
USG abdomen showed excessive gaseous distention of bowel. CT scan abdomen was done which showed dilated stomach and small bowel with mild wall thickening of terminal ileum along with few mildly enlarged iliac and mesenteric lymph nodes. Upper gastrointestinal endoscopy showed dilated stomach and duodenum (megaduodenum) (Figure 2A and 2B). Colonoscopy was normal up to terminal ileum. Cerebrospinal fluid examination showed no cells, with a protein level of 110 mg%, ADA of 2 U/L and sugar of 45 mg% (corresponding blood sugar of 96 mg%). Nerve conduction study andelectromyography was suggestive of demyelinating disease. CT scan of brain showed diffuse white matter hypodensity involving frontal, parietal and occipital cortex without any focal lesion. MRI of brain showed diffuse abnormal T2 white matter hyperintense signals in bilateral hemispheres which appear hypointense on T1, without any post contrast enhancementsuggestive of diffuse severe white matter abnormality in a leukodystrophy pattern with a possibility of mitochondrial cytopathy or any other demyelinating illness (Figure 3).
Serum lactate of the patient was 36.8 mg% (normal range 4.5 – 20 mg%) and serum pyruvate was 0.55 mg% (normal range 0.36 – 0.6 mg%) with lactate / pyruvate ratio of 66.9 (lactate / pyruvate ratio of > 20 is highly suggestive of mitochondrial cytopathy). Electron microscopy of the muscle biopsy showed enlarged mitochondria with vacuolation and blurring of cristae. Thus the diagnosis of mitochondrial cytopathy was made, in the form of MNGIE.
Discussion
Mitochondrial disorders are a diverse group of inherited and acquired disorders characterized by abnormalities in the mitochondria that result in inadequate energy production. Prevalence of established mitochondrial DNA (mtDNA) disease appears to be around 1 in 50 000.[1,2] These disorders often present in early adult life, as multisystem involvement with a progressive disabling neurological syndrome which is responsible for considerable morbidity.[1]
This autosomal recessive disease[3] is uncommon, with < 70 reported patients.[4] The disease is caused by mutations in the gene encoding thymidine phosphorylase (TP), leading to loss of activity of the enzyme.[5] TP is an important factor involved in the control and maintenance of the pyrimidine nucleoside pool of the cell. Defects of TP are thought to produce an excess of dTTP, ultimately affecting both the rate and fidelity of mtDNA replication.[5,6] A chromosomal locus for MNGIE has now beenlocated to 22q13.32-qter, distal to D22S1161, with a maximum two-point LOD score of 6.80 at locus D22S526. [7]
Diagnosis of mitochondrial neuro gastrointestinal encephalopathy (MNGIE) should be considered in view of the prominent gastrointestinal involvement. MNGIE is characterized by external ophthalmoplegia, gastrointestinal dysmotility, cachexia, peripheral neuropathy, and leukoencephalopathy.[8] The disease is relentlessly progressive and typically causes death in early to mid-adulthood. Laboratory abnormalities include serum lactic acidosis (64%), lactate/pyruvate ratio > 20, increased levels of plasma thymidine and deoxyuridine, high CSF protein (89%). EMG shows myogenic changes in 61% while myogenic and
neurogenic in 39%.
Diagnosis is established by muscle biopsy. Mitochondrial changes in muscle fibers are mitochondrial proliferation (ragged red fibers)[9] with COX negative fibers (90%) and SDH positive fibers (60%).[10] Electron microscopy may reveal structural defects in mitochondria in the form of abnormally sized or shaped mitochondria, crystalline inclusions, and proliferation of mitochondria, usually beneath the sarcolemmal membrane. Tertiary laboratory evaluation for mtDNA point mutations and mtDNA southern blot may rarely required.[5,10]
Treatment is largely supportive with special concern of nutrition[8] (including multivitamins and antioxidants). Benefits of coenzyme Q10 treatment may include reduction in lactic acid levels improved muscle strength, and decreased muscle fatigability,[11] but follow up study did not show benefit.[12] Lcarnitine is widely used for treatment, but its role in absence of proven carnitine deficiency remains unresolved.[13] Creatine phosphate therapy is reserved for acute crises as the benefits may be transient.[14] Allogeneic stem cell transplantation may correct biochemical derangements in MNGIE.[5]
References
- Van Coster R, De Meirleir L. Mitochondrial cytopathies and neuromuscular disorders. Acta Neurol Belg. 2000;100:156–61.
- Chinnery PF, Turnbull DM. Clinical features, investigation, and management of patients with defects of mitochondrial DNA. J Neurol Neurosurg Psychiatry. 1997;63:559–63.
- Bardosi A, Creutzfeldt W, DiMauro S, Felgenhauer K, Friede RL, Goebel HH, et al. Myo-, neuro-, gastrointestinal encephalopathy (MNGIE syndrome) due to partial deficiency of cytochrome-c-oxidase. A new mitochondrial multisystem disorder. Acta Neuropathol. 1987;74:248–58.
- Marti R, Spinazzola A, Tadesse S, Nishino I, Nishigaki Y, Hirano M. Definitive diagnosis of mitochondrial neurogastrointestinal encephalomyopathy by biochemical assays. Clinical Chemistry. 2004;50:120–4.
- Hirano M, Marti R, Casali C, Tadesse S, Uldrick T, Fine B, et al. Allogeneic stem cell transplantation corrects biochemical derangements in MNGIE. Neurology. 2006;67:1458–60.
- Hirano M, Nishigaki Y, Marti R. Mitochondrial neurogastrointestinal encephalomyopathy (MNGIE): a disease of two genomes. Neurologist. 2004;10:8–17.
- Hirano M, Garcia-de-Yebenes J, Jones AC, Nishino I, DiMauro S, Carlo JR, et al. Mitochondrial neurogastrointestinal encephalomyopathy syndrome maps to chromosome 22q13.32- qter. Am J Hum Genet. 1998;63:526–33.
- Gillis L, Kaye E. Diagnosis and management of mitochondrial diseases. Pediatr Clin North Am. 2002;49:203–19.
- Teitelbaum JE, Berde CB, Nurko S, Buonomo C, Perez-Atayde AR, Fox VL. Diagnosis and management of MNGIE syndrome in children: case report and review of the literature. J Pediatr Gastroenterol Nutr. 2002;35:377–83.
- Giordano C, Sebastiani M, De Giorgio R, Travaglini C, Tancredi A, Valentino ML, et al. Gastrointestinal dysmotility in mitochondrial neurogastrointestinal encephalomyopathy is caused by mitochondrial DNA depletion. Am J Pathol. 2008;173:1120–8.
- Goda S, Hamada T, Ishimoto S, Kobayashi T, Goto I, Kuroiwa Y. Clinical improvement after administration of coenzyme Q10 in a patient with mitochondrial encephalomyopathy. J Neurol. 1987;234:62–3.
- Matthews PM, Ford B, Dandurand RJ, Eidelman DH, O’Connor D, Sherwin A, et al. Coenzyme Q10 with multiple vitamins is generally ineffective in treatment of mitochondrial disease. Neurology. 1993;43:884–90.
- Pons R, De Vivo DC. Primary and secondary carnitine deficiency syndromes. J Child Neurol. 1995;10:s8–24.
- Tarnopolsky MA, Martin J. Creatine monohydrate increases strength in patients with neuromuscular disease. Neurology. 1999;52:854–7.