48uep6bbphidcol2|ID
48uep6bbphidvals|1838
48uep6bbph|2000F98CTab_Articles|Fulltext
Introduction
Serum Gamma Glutamyl Transpeptidase (GGTP) level is considered a useful screening test for diagnosis of cholestatic liver disorders where it helps in initial differentiation of various disease entities. High GGTP level is seen classically in biliary atresia, progressive familial intrahepatic cholestasis type 3 (PFIC3) and primary sclerosing cholangitis (PSC)etc while low GGTP level is seen in PFIC type 1 & 2 as well as bile acid synthetic defects (BASD) amongst others. However, some patients associated with classically high GGTP cholestasis presenting late in disease course with hepatic synthetic failure, may have progressive fall in GGTP levels. Thus, if these patients are seen at this late stage, they may have low GGTP levels leading to erroneous interpretation. This limitation in diagnostic utility of serum GGTP levels in initial differential diagnosis of advanced cholestatic liver diseases has not been described in literature before. We have, therefore, done this study to assess the diagnostic significance of serum gamma glutamyl transpeptidase (GGTP) in advanced pediatric cholestatic liver diseases with high GGTP cholestasis.
Patients and Methods
Retrospective evaluation of data from August 2011 to May 2016 was done, where all pediatric cases (<18 years age) of cholestatic liver disease presenting to the Department of Pediatric Hepatology of the institute were evaluated, after ethical approval.
Only those cases which fulfilled all the three following criteria were included in the study:
1. High GGTP Cholestasisat baseline (normal ranges for serum GGTP : <2 months: <230 U/L; 2-4 months: <100 U/L; >4 months: < 64 U/L). (modified from Davit-Spraul1).
2. Preserved hepatic synthetic functions at baseline (defined as International Normalised Ratio < 1.3, serum albumin > 3.5 grams/decilitre and no present/past evidence of decompensation of liver disease e.g ascites).
3. Progression of liver disease (> 2 of the following:uncorrectable coagulopathy i.e International Normalised Ratio > 1.5 after 6 hours of vitamin K10 mg injection, serum albumin < 3.5 grams/decilitre and/or present/past evidence of decompensation of liver disease e.g ascites) during study period with at least 1 year follow up.
Neonatal cholestasis syndrome (NCS) was defined by conjugated hyperbilirubinemia as per standard definition.3 In children between 6 months and 18 years of age, cholestatic liver disease was defined asrecurrent or persistent conjugated hyperbilirubinemia with evidence of cholestasis as defined by (any one of following)- clinical (pruritus or acholic stools), biochemical (elevation of alkaline phosphatase (SAP) + GGTP> 3 times of upper limit of normal) and histopathological (bile plugs, cellular and canalicular cholestasis, cholestatic rosettes).2 Biliary atresia(BA) was diagnosed based upon the typical histological features4 with or without ultrasonographic features5 and confirmed on per-operative cholangiography. Diagnosis of PFIC-3 was made on histopathological features and absent MDR3 (multi drug resistant protein-3) antibody immunostaining on liver biopsy. PSC was diagnosed on the basis of classical imaging (multifocal strictures, dilatation and ductal wall irregularity involving the biliary tract, with or without beaded appearance), histopathology (evidence of biliary pathology with cholestasis, ductular reaction and duct damage) or both. These were diagnosed as large duct PSC (positive imaging and biopsy features) or small duct PSC (only biopsy features) after excluding other common etiologies including drug induced injury, structural causes etc. Autoimmune hepatitis/PSC overlap syndrome was also diagnosed based on standard criteria2. The management of these cases was done as per standard disease specific protocol.
Results
Out of a total of 163 cholestatic liver disease children, 134 children (BA = 111, PFIC type 3 = 4, PSC = 10 and PSC/AIH overlap = 9) were diagnosed as having high GGTP cholestasis. Amongst these, 7cases (4 cases of biliary atresia,2 cases of PFIC type 3 and 1 case of PSC) fulfilled the inclusion criteria. The cases are described in detail in Table 1.
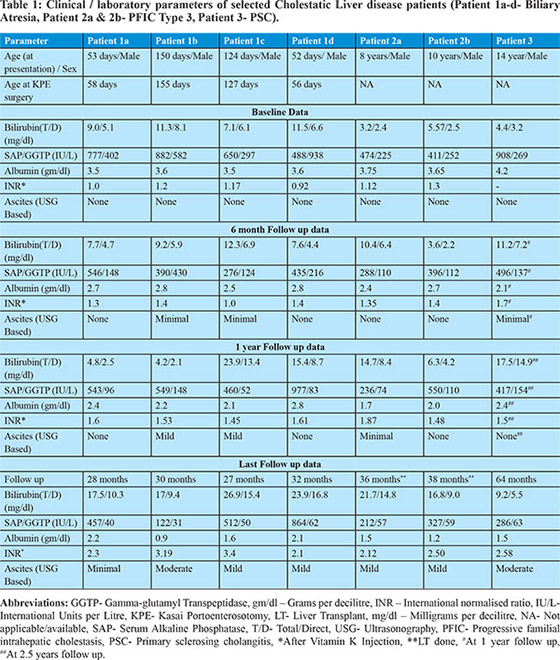
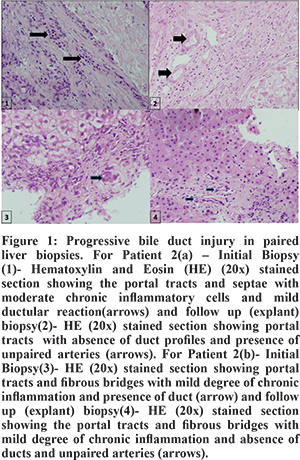
In these cases, we observed that serum GGTP levels initially remained high and gradually decreased as the disease progressedwith decompensation of hepatic function as evident by hypoalbuminemia, worsening coagulopathy and/or ascites [Table1]. The 4 BA cases showed progressive deterioration of hepatic functions after an unsuccessful Kasai portoenterostomy (KPE) surgery (defined as serum total bilirubin > 3 mg/dl 3 months after KPE) as expected from the natural history of the disease. None of these children could be bridged to liver transplantation due to logistic issues and expired later. Two children with PFIC 3(both siblings, patients 2a & 2b) underwent living donor related liver transplant (LDLT)and paired liver biopsies were available for these two patientsduring the course of illness. Over a period of around 3 years, both of them showed progressive bile duct damage with resultant paucity of interlobular bile ducts (Figure 1). One case of PSC (patient 3) had high GGTP cholestasis at baseline with normal synthetic functions. He developed recurrent cholangitis (pain abdomen, jaundice and pruritus) during disease course with each episode characterised by increasing jaundice and transient synthetic failure (hypoalbuminemia,as shown in follow up lab parameters in table 1) with improvement after treatment in each episode. About 5 years after symptom onset, he progressed to end stage liver disease (ascites, hepatic encephalopathy and acute variceal bleed), for which he subsequently underwent LDLT.
Discussion
Ours is the first study to show the progressive fall in serum GGTP levels with advanced liver diseasein cases with high GGTP cholestatic disorders.
Serum GGTP is a microsomal glycoprotein which catalyzes the transfer of ? -glutamyl groups from peptides such as glutathione to other amino acids, and is also involved in hydrolysis of glutathione. It is localized to cell membrane in both hepatocytes and cholangiocytes.6-8 Its significance lies in its diagnostic ability to help differentiate various pediatric cholestatic disorders as stated above. Previous studies have identified different cut offs to differentiate biliary atresia from other causes of neonatal cholestasis.9 Also, recent studies have identified serum GGTP levels as having prognostic importance in cases of infantile cholestasis syndrome.8 In a study by Wang et al, significance of serum GGTP levels was studied in 38 cases with idiopathicneonatal hepatitis. They found that in those cases where GGTP level was high at presentation and then decreased in parallel with their clinicaldeterioration had poor prognosis. They hypothesised that decline of GGTP levels with progressive deranged hepatic function tests represents deterioration of liverfunction.8 The same is true for the present study that all the study cases with progressively falling GGTP levels either died or required LT.
There is usually an over-reliance on serum GGTP levels in considering a differential diagnosis of cholestatic liver disease cases in the developing world where there is a lack of optimum facilities for genetic testing for diagnostic confirmation. This is stark contrast to the western institutions where genetic analysis is the modality of choice. Thus, in scenarios, where patients present late in disease course (with hepatic synthetic failure), which usually is the scenario in developing world, this so called ‘normalisation of GGTP’ during disease course may erroneously point towards a different etiologyi.e low GGTP cholestasis (e.g PFIC type 1 or 2 etc).In such cases, even liver biopsy and subsequent histopathological characterisation is usually not feasible due to coagulopathy and ascites. This is especially important for arriving at an exact diagnosis since we can direct our genetic testing towards a specific suspected disorder avoiding the laborious whole genome sequencing in all cases. If confirmed, this holds significance for future pregnancies and prenatal counselling also. This phenomenon was also highlighted from the fact that in some cases of high GGTP cholestasis (PSC and PSC-AIH overlap) in our study group, serum GGTP levels that were low at presentation (along with hepatic synthetic failure), gradually rose to higher levels after treatment (including ursodeoxycholic acid, immunosuppressive drugs, cholangitis treatment etc as applicable) with partial improvement in hepatic synthetic function (data not shown). Similar mechanism likely occurred in patient 3 with deterioration during cholangitis episodes and partial recovery thereafter.
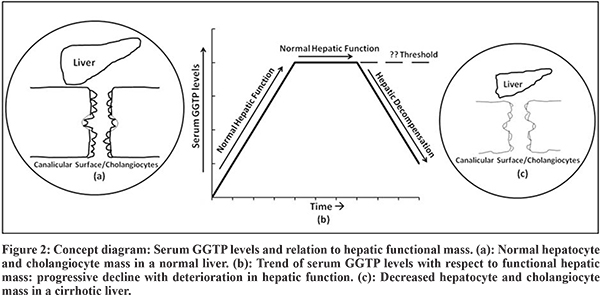
The exact pathophysiology behind this phenomenon of falling GGTP with advancing liver disease remains an enigma. Serum GGTP usually falls slowly after a successful KPE with resolving cholestasis but continues to remain high in those with failure of the surgery till hepatic decompensation occurs. With advancing liver disease, there is likely a loss of functional hepatic mass especially cholangiocytes which are a major source of GGTP [Concept diagram in Figure 2]. In most cases, GGTP levels would start decreasing progressively with decrease in hepatic synthetic function and reach ‘normal’ levels when the liver function goes below threshold which is still undetermined (Table 1). Some patients in the present overall cohort of biliary atresia had shown decreasing GGTP levels (but still in supra-normal range) over the disease course, but were not included in the study cohort due lack of fulfilment of all eligibility criteria (data not shown). Similar evolution has been reported in previous studies on untreated biliary atresia cases where progressive evolution from bile ductular proliferation to duct regression and subsequent progressive intrahepatic duct loss has been documented.10 This likely reflects theprogressive biliary damage occurring in unsuccessful KPE cases and likely other chronic cholestatic liver diseases also.
Ours’ is a tertiary care centre and due to delayed referral from peripheral hospitals, large majority of the patients present late in disease course. Therefore, at our centre, it is uncommon for most liver disease patients to have preserved hepatic function at baseline and this is responsible for small sample size in our study despite a largerbaseline cohort of cholestatic liver disorders. Also, another major limitation of present study is lack of genetic confirmation of PFIC type 3 cases, which is not available in our country.
To conclude, serum GGTP levels should not be used in isolation to consider a differential diagnosis of pediatric cholestatic disorders in resource constraint countries where genetic confirmation is not widely available and should always be used in conjunction with hepatic synthetic functions and clinical picture to avoid misinterpretation.
References
- Davit-Spraul A, Fabre M, Branchereau S, et al. ATP8B1 and ABCB11 analysis in 62 children with normal gamma-glutamyl transferase progressive familial intrahepatic cholestasis (PFIC): phenotypic differences between PFIC1 and PFIC2 and natural history. Hepatology 2010;51: 1645–55.
- EASL Clinical Practice Guidelines: management of cholestatic liver diseases. European Association for the Study of the Liver. J Hepatol. 2009 Aug;51(2):237-67
- Guideline for the Evaluation of Cholestatic Jaundice in Infants: Recommendations of the North American Society for Pediatric Gastroenterology, Hepatology and Nutrition. Journal of Pediatric Gastroenterology and Nutrition. August 2004; 39(2):115–128
- Lee WS, Looi LM. Usefulness of a scoring system in the interpretation of histology in neonatal cholestasis. World J. Gastroenterol. 2009; 15: 5326–33.
- Humphrey TM, Stringer MD. Biliary atresia: US diagnosis. Radiology 2007; 244:845–51.
- Cabrera-Abreu JC, Green A. Gamma-glutamyltransferase: value of its measurement in paediatrics.Ann ClinBiochem. 2002 Jan;39(Pt 1):22-5.
- Herlong H.L, Mitchell Jr M.C. Laboratory tests. In: Schiff ER, Maddrey WC, Sorrell MF, eds. Schiff’s diseases of the liver. 11th ed. Sussex: Wiley-Blackwell, 2012;17-43.
- Wang JS, Wang ZL, Wang XH, Zhu QR, Zheng S. The Prognostic Value of Serum Gamma Glutamyltransferase Activity in Chinese Infants with Previously Diagnosed Idiopathic Neonatal Hepatitis. HK J Paediatr (New Series) 2008;13:3945.
- Tang KS, Huang LT, Huang YH, Lai CY, Wu CH, Wang SM, Hwang KP, Huang FC, Tiao MM.Gamma-glutamyl transferase in the diagnosis of biliary atresia. Acta Paediatr Taiwan. 2007 Jul-Aug;48(4):196-200.
- Landing BH, Wells TR, Ramicone E. Time course of the intrahepatic lesion of extrahepatic biliary atresia: a morphometric study. Pediatr Pathol. 1985;4(3-4):309-19.