48uep6bbphidcol2|ID
48uep6bbphidvals|2945
48uep6bbph|2000F98CTab_Articles|Fulltext
Autoimmune pancreatitis was first described in 1961 by Sarles et al. as a particular form of chronic pancreatitis that was associated with hypergammaglobulinemia.1 The term “AIP” was first used by Yoshida et al. to describe a corticosteroid-responsive disease with features of autoimmunity.2 In 2011, the International Association of Pancreatology proposed International Consensus Diagnostic Criteria (ICDC), which is composed of five cardinal features including pancreas histology and imaging findings, serology, presence of other organ involvement and prompt response to corticosteroids. AIP has been divided into type 1 or type 2 AIP. A high prevalence of type 1 AIP is observed in Asia, whereas type 2 mainly occurs in Western countries. At present paediatric gastroenterologists rely on the adult AIP guidelines to diagnose and manage AIP in children. However, as the clinical presentation is different in the paediatric population, the exclusive use of adult criteria may lead to underdiagnosis of AIP in children.3 This case report describes the clinical and radiological presentation of AIP in a five-year-old boy.
Case Report
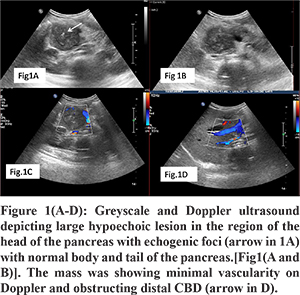
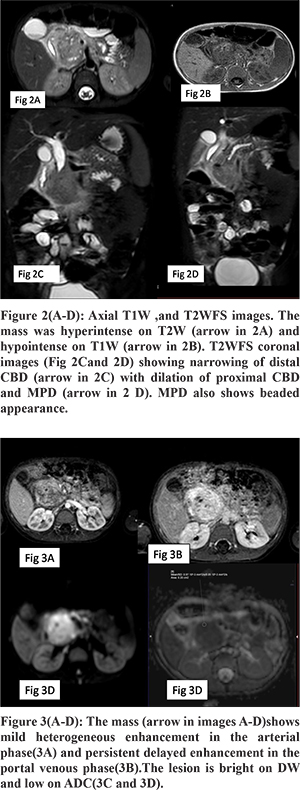
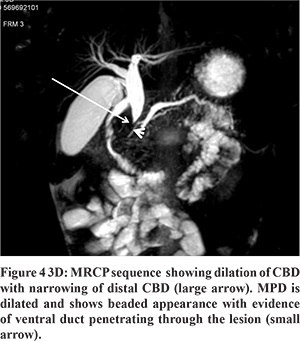
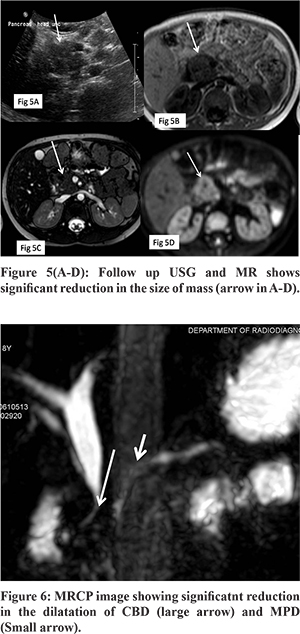
A 5-year-old boy presented with a two months history of abdominal pain, fever, and weight loss. The abdominal pain was epigastric in location, dull aching in nature, not related to meals, and associated with documented high-grade fever. Blood analysis revealed bilirubin 1.4 mg/dL (normal 0.2-1.2 mg/dL), Aspartate aminotransferase 163 U/L (normal 5-40 U/L), Alanine aminotransferase 80U/L (normal 5-45 U/L). Another fever workup, including malarial smears, was negative; and blood culture was sterile. To evaluate for the cause of abdominal pain, an ultrasound abdomen was done, which revealed a large lobulated hypoechoic mass lesion in the region of the head of the pancreas with specks of calcification and dilatation of the common bile duct and main pancreatic duct(MPD) in the region of body and tail. The mass was showing minimal vascularity on color Doppler (Figure 1A-D). On MR with MRCP, the mass was heterogeneously hyperintense on T2W and hypointense on T1W images (Figure 2A and 2B) and was causing narrowing of distal CBD (Figure 2C). MPD was dilated in the pancreatic neck, body, and tail and had a beaded appearance. Ventral PD was seen coursing through the lesion (Duct penetrating sign positive) (Figure 2D and 4). On the dynamic contrast-enhanced sequence, the mass showed mild heterogeneous enhancement in the arterial phase, progressive increase in the Portal venous, and delayed phase (Figure 3A and B). On DWI, the mass showed marked restriction (ADC value -0.97x103) (Figure 3C and D). Serum IgG4 levels were normal. The imaging findings were not conclusive of any specific diagnosis, and malignancy could not be excluded. Hence US-guided biopsy was performed, which revealed extensive fibrosis infiltrated with moderate to dense mixed inflammation including few plasma cells and few eosinophils. However, the storiform pattern of arrangement and phlebitis was not identified in the biopsy, and stain for IgG4 was negative. Because of no malignant features on biopsy, we decided to give a trial of steroids suspecting autoimmune pancreatitis despite classical described features being not present. After six weeks follow up, ultrasound was performed, which showed a significant reduction in the size of mass (Figure 5A-D). MRCP also revealed a reduction in the dilation of CBD and MPD. Distal CBD was seen; however, it was narrowed in calibre (Figure 6). The child was put on a gradually tapering course of steroids, and azathioprine was added.
Discussion
Autoimmune pancreatitis (AIP) refers to a unique form of steroid-responsive chronic pancreatitis. As defined by ICDC, AIP is characterized by frequent presentation with obstructive jaundice with or without pancreatic mass, histologically by a lymphoplasmacytic infiltrate and fibrosis and therapeutically by a dramatic response to steroids.
AIP in adults is classified into two subgroups; with differences in epidemiology, clinical profile, histopathology, natural history, and outcome. Type 1 AIP is called lymphoplasmacytic sclerosing pancreatitis(LPSP) or AIP without granulocyte epithelial lesions (GELs). Clinically this type appears to be the pancreatic manifestation of IgG4-related systemic disease characterized by elevated serum IgG4 levels and extra pancreatic lesions (e.g., sclerosing cholangitis, sclerosing sialadenitis and retroperitoneal fibrosis). Histopathologically it is characterized by infiltration with abundant IgG4-positive plasma cells. Another type is Idiopathic Duct-centric pancreatitis (IDCP) or AIP with GEL, i.e., type 2 AIP. Idiopathic duct-centric pancreatitis usually has none or very few (<10 cells/HPF) IgG4-positive plasma cells and is not associated with elevated serum IgG4 or other systemic involvement as seen in LPSP. As there is no serological marker for type 2 AIP, and it lacks organ involvement, definitive diagnosis requires pancreatic histopathology.
AIP is a well-described disease in adults, but in children, most of the information relies on case reports and case series. Scheers et al. in 2017 reviewed and reported analysis for 30 paediatric AIP cases from 19 publications, also enrolled 18 patients diagnosed as AIP in INSPIRE and CUSL databases. They proposed that a diagnosis of AIP in children can be established based on the combination of specific clinical symptoms at presentation and imaging findings in contrast to adults.4
The most frequent clinical presentation of AIP is obstructive jaundice or a pancreatic mass, though sometimes it is asymptomatic. Early imaging findings are pancreatic enlargement with or without mass. Late findings include pancreatic atrophy, calcification, ductal dilatation. When presents as a pancreatic mass/focal enlargement, it is a diagnostic challenge to distinguish it from a neoplastic condition such as pancreatoblastoma or solid pseudopapillary epithelial neoplasms that may be encapsulated with cystic or solid components.
The histopathological pattern of LPSP shows four characteristic features, (1) dense infiltration of plasma cells and lymphocytes, particularly periductal, (2) peculiar storiform fibrosis, (3) venulitis with lymphocytes and plasma cells often leading to obliteration of affected veins, and (4) abundant (>10 cells per HPF) immunoglobulin IgG4 positive plasma cells. Idiopathic duct-centric pancreatitis and LPSP share some histopathological features, such as periductal lymphoplasmacytic infiltrate and storiform fibrosis. A characteristic feature of IDCP, not seen in LPSP, is the GEL: intraluminal and intraepithelial neutrophils in medium-sized and small ducts as well as acini, often leading to the destruction and obliteration of duct lumen.4
On imaging, AIP can either present as diffuse pancreatic enlargement or focal mass. A diffuse type of AIP is more common in adults. Characteristic imaging findings seen on magnetic resonance imaging, include hypo intensity of the parenchyma on T1-weighted images, slight hyperintensity on T2-weighted images, and delayed enhancement during the late phases of contrast.5 On T2W MRI, a hypointense rim(halo sign) is highly specific for AIP but is seen in only 30% to 40% of patients at diagnosis.5 The focal mass forming pancreatitis is more commonly seen in children,4 which has to be differentiated from the malignancy. Various differentiating points on MRI have been described to differentiate mass-forming AIP from pancreatic adenocarcinoma, including homogeneous enhancement, delayed enhancement in portal venous phase, duct penetrating sign, and low ADC value less than 0.90x10-3mm2.6 In our case, all these imaging findings were seen, which favoured the diagnosis of AIP. A detailed description of the pancreas and ductal pathology is challenged by limitations of the MRCP resolution in young children and less frequent use of ERCP compared with adults.7
Serum IgG4 is the single best marker for AIP. Approximately two-thirds of adult patients with type 1 AIP and 23% of patients with type 2 AIP have elevated IgG4 levels.4 In contrast, IgG4 serology and staining are rarely positive in children with AIP. As suggested by Scheers et al., it is possible that children more commonly follow the disease presentation of AIP type 2 or have a distinct AIP pattern that may not fall into either category. Nevertheless, the histology of pancreatic biopsies, as described in adult guidelines4, has not been validated for the disease phenotype in children. Thus the clear distinction of subgroups is difficult in children.
AIP is a fibroinflammatory disease and, in the early phase, is characterized by robust inflammation. The therapies target inflammatory response to provide relief of symptoms and decrease or delay the progression to fibrosis. Till now, there are no randomized controlled trials of therapy in AIP. However, based on observational data, corticosteroids are the mainstay of treatment. Furthermore, for patients who are either intolerant of high-dose corticosteroids or have multiple relapses despite therapy, there are emerging data to support the use of other treatment options, including corticosteroid-sparing immunomodulators and B-cell depletion therapy using rituximab.8
Generally, patients with AIP show an excellent response to corticosteroids, clinical as well as radiological. Thus a steroid trial of up to two weeks is a part of the diagnostic criteria for adult AIP. Most paediatric studies demonstrate a clear response of up to 92% 4 of AIP to steroids. After remission, many regimens include a slow, prolonged taper over several months to a low maintenance dose continued for 1 to 3 years and, in some instances, indefinitely.8
Conclusion
The reported case is interesting as it did not meet all the criteria for AIP, and had an unusual presentation, suspecting it clinically with a radiological diagnosis. Definite response to steroids further supported the diagnosis. It does emphasize that presentation in children is varied, and diagnosis as per adult criteria is difficult.
References
- Sarles H, Sarles J-C, Muratore R, Guien C. Chronic inflammatory sclerosis of the pancreas—An autonomous pancreatic disease? Am J Dig Dis. 1961 Jul 1;6(7):688–98.
- Yoshida K, Toki F, Takeuchi T, Watanabe S-I, Shiratori K, Hayashi N. Chronic pancreatitis caused by an autoimmune abnormality. Dig Dis Sci. 1995 Jul 1;40(7):1561–8.
- Shimosegawa T, Chari ST, Kawa S, Mino-Kenudson M, Kim M-H, Kloppel G, et al. International Consensus Diagnostic Criteria for Autoimmune Pancreatitis. 2011;40(3):7.
- Scheers I, Palermo JJ, Freedman S, Wilschanski M, Shah U, Abu-El-Haija M, et al. Autoimmune Pancreatitis in Children: Characteristic Features, Diagnosis, and Management. Am J Gastroenterol. 2017 Oct 1;112(10):1604–11.
- Bodily KD, Takahashi N, Fletcher JG, Fidler JL, Hough DM, Kawashima A, et al. Autoimmune Pancreatitis: Pancreatic and Extrapancreatic Imaging Findings. Am J Roentgenol. 2009 Feb 1;192(2):431–7.
- Choi S-Y, Kim SH, Kang TW, Song KD, Park HJ, Choi Y-H. Differentiating Mass-Forming Autoimmune Pancreatitis From Pancreatic Ductal Adenocarcinoma on the Basis of Contrast-Enhanced MRI and DWI Findings. Am J Roentgenol. 2016 Jan 21;206(2):291–300.
- Chavhan GB, Babyn PS, Manson D, Vidarsson L. Pediatric MR Cholangiopancreatography: Principles, Technique, and Clinical Applications. RadioGraphics. 2008 Nov 1;28(7):1951–62.
- Ghazale A, Chari ST, Zhang L, Smyrk TC, Takahashi N, Levy MJ, et al. Immunoglobulin G4–Associated Cholangitis: Clinical Profile and Response to Therapy. Gastroenterology. 2008 Mar;134(3):706–15.