48uep6bbphidcol2|ID
48uep6bbphidvals|1875
48uep6bbph|2000F98CTab_Articles|Fulltext
Hepatic Encephalopathy (HE) may be the presenting manifestation of portosystemic shunting of blood, which usually occurs in the setting of chronic liver disease with portal hypertension. Very rarely, congenital portosystemic shunt (CPSS) may present with HE. We present a 47 year old man, who on evaluation was found to have a CPSS, also called Abernethy Malformation (Type 2B). The shunt resulted in hyperammonemia and a steal phenomenon causing hepatic hypoperfusion, both of which were corrected after the shunt was successfully ligated surgically.
Case Report
A 47 years old gentleman, presented to our gastroenterology services with prior history of two episodes of altered sensorium lasting for one week each. First episode was 8 months back when he was on a mountaineering vacation in Nepal at 22,000 feet above sea level, where his oxygen saturation was 88%. He developed insidious onset irritability, confusion followed by coma requiring hospitalization. At this time, on direct questioning, he also had constipation for 7 days. His routine blood workup was normal, except for a low platelet count. Cerebrospinal fluid analysis was normal and electroencephalography showed diffuse cerebral dysfunction with burst suppression pattern. Diagnosis of high altitude cerebral edema was suggested and patient treated with intravenous mannitol and dexamethasone. His sensorium gradually and completely improved to normal. He remained asymptomatic until one month back, when he developed similar episode of altered sensorium requiring hospitalization. Preceding this episode, he described increased work related stress for about 5 days, excess non-vegetarian food intake and constipation. He was found to have high blood ammonia levels of 315 µg/100 ml (25-94 µg/100 ml) which decreased to 107 µg/100 ml after treatment with lactulose and rifaximin. Screening ultrasound abdomen and liver function tests were normal. MRI of the abdomen showed a normal liver and normal portal vein with attenuated intra-hepatic portal venous radicles, and a large tortous vein in the retroperitoneum connecting the portal circulation to the left renal vein (Figure 1).
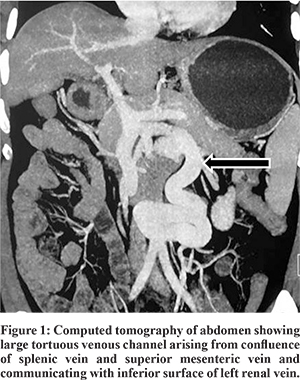
At presentation to us, he had no jaundice or any signs of chronic liver disease. He had grade 2 clubbing. He completed number connection test (NCT) in 12 sec. His work up showed mild pancytopenia, normal liver function tests, coagulation profile and no varices on esophagogastroduodenoscopy. There was no orthodeoxia, and contrast echocardiography did not show any evidence of hepatopulmonary syndrome (HPS). Triphasic CECT abdomen showed a portosystemic shunt of 17 mm diameter arising from the PV confluence, coursing inferiorly to take a ‘U’ shaped turn and communicating with the left renal vein (Type 2B Abernethy malformation). Liver was 13.6 cm with mildly irregular outline with spleen 13.7 cm.
Doppler study showed a shunt velocity of 22 cm/sec. Transient elastography (FibroScan; Echosens, Paris, France) showed mildly increased liver stiffness of 12 kPa.
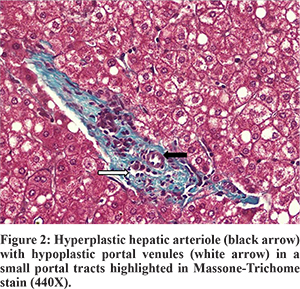
A diagnosis of Type-2 Abernethy syndrome with recurrent HE and hypersplenism was made. He underwent a single step shunt ligation, splenectomy and liver biopsy. At surgery, the liver was pink, reduced in size with no evidence of cirrhosis. A large shunt joining the splenoportal confluence to the left renal vein was identified and clamped. Intraoperative Doppler showed good portal blood flow after clamping and thus the shunt was ligated. Postoperative doppler showed improved intrahepatic and extrahepatic portal blood flow. His liver biopsy showed prominent hepatic arterial branches, normal portal venules, fatty change, and presence of centrocentral fibrosis (Figure 2). There was no evidence of nodular regenerative hyperplasia on biopsy. Despite use of octreotide to avoid anticipated acute rise in portal pressures, he developed transient ascites. His arterial ammonia level normalized (38 µg/100 mL), NCT improved (10 sec) and platelets increased to 153,000/mm3. He is asymptomatic on follow up at three months.
Discussion
Abernethy malformation is an extremely rare congenital abnormality and only about 101 cases have been reported since its first description in 1793 by John Abernethy.1,2 It was further classified into Type 1 shunt in which all the portal venous blood completely bypasses the liver, and type 2 shunt in which, there is partial shunting of portal flow into systemic circulation by a side-to-side shunt (Table 1).3,4 CPSS often have a complex and varied anatomy, and arise due to abnormal persistence of anastomotic channels between the vitelline and subcardinal venous system.
CPSS is associated with reduced blood flow to liver with resultant reduced liver volume, decreased hepatic glycogen and regenerative capacity. There is compensatory hypertrophy of the hepatic artery to maintain hepatic perfusion, thus liver is pink in color than its usual purple. This also increases the risk of hepatic tumors such as hemangiomatosis, hepatoblastoma, and hepatocellular carcinoma.
Type 2 Abernethy syndrome may be asymptomatic or may present in childhood with liver dysfunction, HPS, congenital abnormalities such as cardiac defects (60%), biliary atresia (20%), polysplenia (20%), and malrotation (10%), or rarely HE, pyschiatric disturbances and mental retardation.5 HE is often seen among adults due to the effect of ammonia on an aging brain and is usually precipitated by GI bleed, constipation, systemic infection and possibly hypoxia, which likely operates by direct neuronal inhibition.6 Liver dysfunction manifests as fatty change, increased fibrosis and decreased liver volume, as seen in our patient. Rarely liver tumors, chronic liver dysfunction and cirrhosis have been reported. Absence of portal hypertension is an important hallmark of CPSS.
Therapeutic options are dependent on the type of shunt, shunt fraction, age of patient, presence or absence of other congenital abnormalities, and nature of clinical presentation. Type-2 shunts can be managed by a single stage surgical ligation or radiological intervention. Shunt ligation restores portal flow, reduces encephalopathy and risk of hepatic tumors.7,8 Postoperatively, patients may develop transient portal hypertension which may manifest as ascites and UGI bleed, and octreotide may be helpful.
Conclusion
Diagnosis of CPSS requires a high index of suspicion. Unexplained encephalopathy with elevated blood ammonia levels in the absence of portal hypertension or liver dysfunction should initiate a radiological search for a congenital shunt. Ligation of the shunt results in restoration of normal portal circulation, complete resolution of symptoms and decreased risk of tumor formation in the longterm.
References
- Abernethy J. Account of two instances of uncom¬mon formation in the viscera of the human body. Philos Trans R Soc Lond 1793;17:292–9.
- HAO Y, HONG X, ZHAO X. Congenital absence of the portal vein associated with focal nodular hyperplasia of the liver and congenital heart disease (Abernethy malformation): A case report and literature review. Oncology Letters. 2015;9:695-700.
- Howard ER, Davenport M. Congenital extrahepatic portocaval shunts: the Abernethy malformation. J Pediatr Surg 1997;32:494–7.
- Morgan G, Superina R. Congenital absence of the portal vein: two cases and a proposed classification system for portasystemic vascular anomalies. J Pedi¬atr Surg 1994;29:1239–41.
- Murray CP, Yoo SJ, Babyn PS. Congenital extrahe¬patic portosystemic shunts. Pediatr Radiol 2003;33:614–20.
- Emre S, Arnon R, Cohen E, Morotti RA, Vaysman D, Shneider BL. Resolution of hepatopulmonary syndrome after auxiliary partial orthotropic liver transplantation in Abernethy malformation: a case report. Liver Transpl 2007;13:1662-8.
- Gallego C, Miralles M, Marin C, Muyor P, Gonzales G, Garcia-Hidalgo E. Congenital hepatic shunts. RadioGraphics 2004;24:755-72
- Blanc T, Guerin F, Franchi- Abella S et al. Congenital portosystemic shunts in children ; A new anatomical classification correlated with surgical strategy. Ann Surg 2014;260:188-98.