|
Thoguluva Seshadri Chandrasekar, Kalamegam Raja Yogesh, Bollu Janakan Gokul, Suriyanarayanan Sathiamoorthy, Menta Sanjeevaraya Prasad, Thoguluva Chandrasekar Viveksandeep Med India Hospitals, Chennai
Corresponding Author:
Dr Thoguluva Seshadri Chandrasekar Email: tscmedindia@yahoo.com
DOI:
http://dx.doi.org/10.7869/tg.476
48uep6bbphidcol2|ID 48uep6bbphidvals|1859 48uep6bbph|2000F98CTab_Articles|Fulltext Hereditary Haemorrhagic telangiectasia (HHT), also known as Osler-Weber-Rendu syndrome is an autosomal dominant disorder, characterized by the presence of multiple mucocutaneous telangiectasias, visceral arteriovenous malformations, gastrointestinal bleeding and iron deficiency anaemia. The prevalence ranges from 1 in 5000 to 1 in 10,000.1 Mutations of the endoglin (ENG) and activin receptor–like kinase 1 (ALK-1) genes have been implicated in its pathogenesis.2 The condition is under-recognized and the diagnosis, often delayed. The presentation is varied, and accordingly, patients report to a wide range of clinical specialties.The diagnosis is established based on the presence of any three of the four Cura?ao Criteria3 which includes 1) Epistaxis 2) Telangiectasias 3) Visceral arteriovenous malformations and 4) Family history of affected first degree relative. Early recognition of this condition is vital to offer appropriate treatment, thereby prevent life-threatening complications. Screening of family members should also be emphasized to prevent adverse outcomes. Here we report the case of a middle-aged lady who presented with chronic iron deficiency anaemia secondary to occult gastrointestinal bleed, in whom the diagnosis of Hereditary haemorrhagic telangiectasia was made.
Case Report
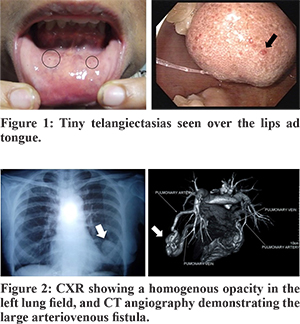
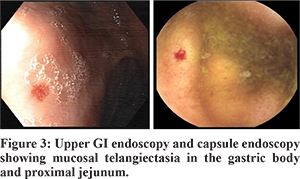
A 54-year-old lady presented with the history of exertional dyspnea and fatigue over the past two years. She denied any history of hematemesis, melena or haematochezia. Her past history was not contributory except for multiple episodes of epistaxis since her childhood. Before she presented to us, she had been evaluated for iron deficiency anaemia and had received multiple blood transfusions. General physical examination was remarkable for pallor and the presence of multiple tiny to small blanchable reddish lesions over the tongue, lips and buccal mucosa (Figure 1). There was no cyanosis or clubbing. On auscultation, a continuous murmur of grade 4/6 was heard over the left infra-scapular region. The other systems were unremarkable. The haemogram showed a haemoglobin of 72 g/L (N: 120 to 155 g/L), haematocrit of 24 % (N: 36 to 46 %), and a mean corpuscular volume (MCV) of 69 fL (N: 78 to 102 fL). The serum iron indices were suggestive of iron deficiency. The total leucocyte, differential and platelet counts, and coagulation profile were within normal limits. The stool examination was positive for occult blood. The chest X-ray showed a well-defined rounded homogenous opacity in the left lower zone (Figure 2). The CT angiography of thorax revealed a large arteriovenous fistula in the left lung (Figure 2). Screening for the presence of other visceral lesions revealed small cerebral and splenic arteriovenous malformations. The upper GI endoscopy, capsule endoscopy and colonoscopy revealed multiple telangiectasias in the stomach, duodenum, jejunum and rectum (Figure 3). The diagnosis of Hereditary Haemorrhagic Telangiectasia was thus made based on the Cura?ao Criteria.
The telangiectasias were ablated endoscopically by Argon plasma coagulation and she was subsequently started on oral haematinics. The large pulmonary arteriovenous malformation was managed angiographically by coil embolization. On follow up, her hemoglobin levels showed a steady rise and five months later, it had risen from the initial 72 gm/L to 108 gm/L. Discussion
Bleeding vascular malformations constitute 6% of cases of GI bleeding.4 Fifteen percent of patients with HHT present with GI bleeding.5 The incidence of gastrointestinal bleeding increases with age with more than 25% of patients above 60 years presenting with melena.6 The lack of elastic lamina or muscle tissue prevents reflex vasoconstriction resulting in prolonged bleeding and chronic blood loss.Though the telangiectasias are distributed throughout the GI tract, the ones in the stomach, duodenum and proximal jejunum tend to bleed more often.7 Such bleeding from telangiectasias can be successfully controlled by endoscopic argon plasma coagulation or thermal contact coagulation. Hormonal therapy with oestrogen and progesterone has also been used as a treatment modality for diffuse small bowel telangiectasias.8 The larger visceral arteriovenous malformation of the lung, central nervous system and liver are often symptomatically silent but, can present with more serious complications such as high-output cardiac failure, massive haemoptysis, pulmonary hypertension, embolic stroke, intracranial haemorrhage and cerebral abscess. Hence they require prophylactic angiographic embolization or surgical resection.
References - Kjeldsen A, Vase P, Green A. Hereditary haemorrhagic telangiectasia: a population-based study of prevalence and mortality in Danish patients. J Intern Med. 1999;245(1):31-39.
- Azuma H. Genetic and molecular pathogenesis of hereditary hemorrhagic telangiectasia. The Journal of Medical Investigation •. 2000;47(3-4)(September 2000):81-90.
- Shovlin C, Guttmacher A, Buscarini E, Faughnan M, Hyland R, Westermann C et al. Diagnostic criteria for hereditary hemorrhagic telangiectasia (Rendu-Osler-Weber syndrome). American Journal of Medical Genetics. 2000;91(1):66-67.
- Sleisenger, M. H., Feldman, M., Friedman, L. S., & Brandt, L. J. (2010). Sleisenger and Fordtran’s gastrointestinal and liver disease: Pathophysiology, diagnosis, management. Philadelphia, PA: Saunders/Elsevier. Vol 1 Pg. 293
- Begbie M. Hereditary haemorrhagic telangiectasia (Osler-Weber-Rendu syndrome): a view from the 21st century. Postgraduate Medical Journal. 2003;79(927):18-24.
- Kjeldsen A. Gastrointestinal bleeding in patients with hereditary hemorrhagic telangiectasia. The American Journal of Gastroenterology. 2000;95(2):415-418.
- Chamberlain S, Patel J, Carter Balart J, Gossage Jr J, Sridhar S. Evaluation of patients with hereditary hemorrhagic telangiectasia with video capsule endoscopy: a single-center prospective study. Endoscopy. 2007;39(6):516-520.
- van Cutsem E, Rutgeerts P, Vantrappen G. Treatment of bleeding gastrointestinal vascular malformations with oestrogen-progesterone. The Lancet. 1990;335(8695):953-955.
|