48uep6bbphidcol2|ID
48uep6bbphidvals|1799
48uep6bbph|2000F98CTab_Articles|Fulltext
Tyrosinemia type 1(Tyr-1) is an autosomal recessive disease of the tyrosine pathway in which the enzyme Fumarylacetoacetate Hydrolase (FAH) is deficient.
1 This can lead to accumulation of mutagenic metabolites leading to the development of cirrhosis of liver and Hepatocellular Carcinoma (HCC).
Case Report
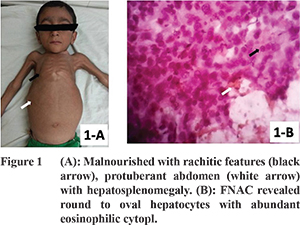
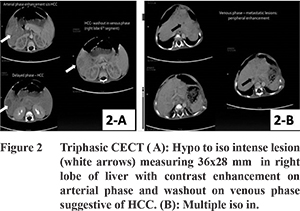
A 4 year old male child, product of consanguineous marriage, presented with failure to thrive, distension of abdomen, and generalized weakness and was diagnosed as Tyr-1 at 1½ years. Patient’s siblings were investigated and found to be normal. Patient was prescribed dietary therapy and Nitisinone, but was noncompliant due to financial reasons. He presented with worsening of above symptoms since the past 3 months along with upper abdominal pain, loss of appetite and significant loss of weight. On examination patient was malnourished with a BMI of 12, with rachitic features (Figure 1A), protuberant abdomen with hepatosplenomegaly, renomegaly, and motor weakness. In view of his recent gross loss of appetite and weight, and abdominal pain, he was investigated for progression to HCC. Serum investigations revealed increased tyrosine levels of 445 micromoles/L, alkaline phosphatase of 1184 U/L, AFP levels of 30285 ng/ml, and decreased serum phosphate levels of 3.7 mg/dL. The urine metabolic screen was positive for aminoaciduria, and the urine organic acid profile showed a 70 fold elevated SA levels. Abdominal ultrasonography detected hepatomegaly with cirrhosis, multiple liver nodules within right lobe of liver suggestive of HCC, splenomegaly and bilateral renomegaly. Triphasic CECT (Figures 2A, 2B) revealed a hypo to iso-intense lesion measuring 36x28 mm in Right lobe of liver (segment 6) with contrast enhancement on arterial phase and washout on venous phase suggestive of HCC, and multiple iso-intense lesions in both lobes of liver with minimal enhancement on arterial phase, but enhancement noted in venous phase suggestive of metastases FNAC of liver lesions (Figure 1B) round to oval abundant eosinophilic cytoplasm with vesicular nuclei, intranuclear inclusions, pleomorphism, prominent nucleoli and bile plugs were found, confirming HCC. The patient’s performance status was 3, hence he was in BCLC Stage “D” HCC for which LT is not indicated. Hence, only symptomatic treatment was given.
Discussion
Tyr-1 typically manifests in early infancy as an acute hepatic crisis with hepatomegaly and bleeding diathesis, which may resolve spontaneously, or persist with failure to thrive.
1 Tyr-1 can also present post-infancy as a chronic form which manifests as Fanconi syndrome, rickets, cardiomyopathy, hepatic manifestations such as acute hepatitis, acute liver failure and cirrhosis.
2 Tyr-1 can lead to HCC, as the pathophysiology involves accumulation of Fumarylacetoacetate (FAA), Maleylacetoacetate (MAA), and Succinylacetone (SA), which being alkylating agents lead to oxidative stress, nucleic acid alkylation, and inhibition of DNA ligase activity - thus leading to DNA and RNA damage and mutagenesis.
3 In Tyr-1, HCC can present with a locally advanced stage or with metastatic disease, with dismal 5-year survival rates ranging from 0% to 9%.
4 Our patient’s presentation was the post infancy chronic type of Tyr-1 with Rickets, Fanconi syndrome, raised AFP and progressive liver disease. Nitisinoneis effective inpreventing the formation of toxic compounds FAA, SA, and SAA and is helpful in controlling liver failure in 90% of patients and in preventing progression to HCC.5 Nitisinone has no role once HCC development occurs.
5
References
- Shah I. Tyrosinemia: a report of three cases from India. Indian J Gastroenterol. 2013;32(2):123-126.
- Castilloux J, Laberge A, Martin S, Lallier M, Marchand V. “Silent” Tyrosinemia Presenting as Hepatocellular Carcinoma in a 10-year-old Girl. J Pediatr Gastroenterol Nutr. 2007;44(3):375-377.
- Prieto-Alamo M, Laval F. Deficient DNA-ligase activity in the metabolic disease tyrosinemia type I. Proc Natl Acad Sci U S A. 1998;95(21):12614-12618.
- Katzenstein H. Hepatocellular Carcinoma in Children and Adolescents: Results From the Pediatric Oncology Group and the Children’s Cancer Group Intergroup Study. J Clin Oncol. 2002;20(12):2789-2797.
- McKiernan P. Nitisinone in the Treatment of Hereditary Tyrosinaemia Type 1. Drugs. 2006;66(6):743-750.