|
|
|
|
 |
|
| |
 |
|
|
Case Report |
|
|
|
|
|
Keywords :
|
|
|
Rishi Bolia, Anshu Srivastava, Ujjal Poddar, Surender Kumar Yachha
Department of Paediatric Gastroenterology,
Sanjay Gandhi Postgraduate Institute of Medical Sciences,
Lucknow, India
Corresponding Author:
Dr. Anshu Srivastava
Email: avanianshu@yahoo.com
DOI:
http://dx.doi.org/10.7869/tg.302
48uep6bbphidvals|1366 48uep6bbphidcol2|ID 48uep6bbph|2000F98CTab_Articles|Fulltext Acute intermittent porphyria (AIP) is the most common hepatic porphyria. However, diagnosis of AIP is often delayed as its manifestations are variable and non-specific. AIP is a disorder of the third or fourth decade, it rarely manifests before puberty. Here we report two cases of AIP who presented in pre pubertal age.
Case 1
A 9-year old boy presented to the outpatient services with a 2-year history of recurrent abdominal pain. The pain was periumblical, episodic, moderate in intensity, once every 2-3 months, lasting for a few hours. There was no history of abdominal distension, vomiting or constipation. Physical examination was unremarkable with normal growth parameters. The child was evaluated elsewhere outside our hospital with complete blood counts, serum amylase, x–ray abdomen, urine and stool examination, abdominal ultrasound and a CECT abdomen. All these investigations were reported normal. The child was suspected to have functional abdominal pain possibly abdominal migraine as the episodes were intense, paroxysmal and intermittent with pain-free periods between the episodes. After one more visit to our outpatient department a month later,the child was lost to follow-up.
Six months later he presented to the emergency with seizures. At presentation he was in a post–ictal state, with a Glasgow coma score of9/15(E3M4V2), heart rate of 134/minute and BP of 174/110 mm Hg. There was no focal deficit and meningeal signs were absent. Fundus examination was normal. The abdomen was slightly distended with sluggish bowel sounds. There was no organomegaly, tenderness or guarding and liver dullness was preserved. Sexual maturity rating was pre–pubertal and there were no other findings on general physical and systemic examination.
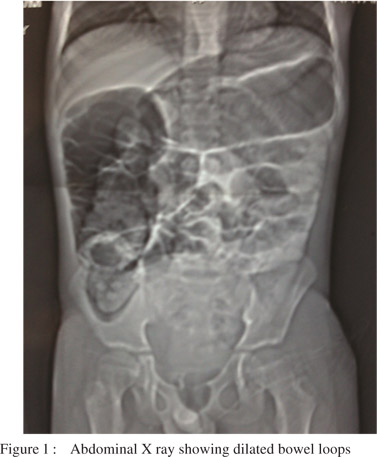
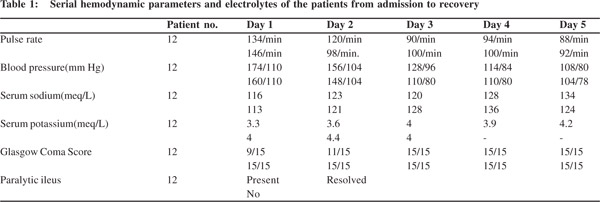
Laboratory investigations showed hyponatremia (serum sodium 116 meq/L) and hypokalemia (serum potassium 3.3 meq/L).Serum calcium, magnesium and blood sugars were normal. A CT head and CSF examination done elsewhere were unremarkable. Plain X–ray abdomen (erect) showed dilated bowel loops (Figure 1). Urine for porphobilinogen was positive. Thus a diagnosis of AIP was made. The child was started on 10% dextrose infusion. For control of BP, nitroglycerine infusion (1 mcg/kg/min and increased upto 5 mcg/kg/min), atenolol(1mg/kg/day) and prazosin (0.5 mg/kg/day) were given. The child’s BP fell with therapy (Table 1) and nitroglycerine infusion was tapered and stopped over 48 hours. Thereafter, the other anti-hypertensive agents were gradually reduced and stopped based on the blood pressure recordings.
The echocardiogram was normal. For the hyponatremia, the child was given a bolus of 3% saline (5 mL/kg), after which normal saline was administered. Sodium levels gradually improved over 3 days.
For seizures, gabapentin was started (15 mg/kg/day PO in 3 divided doses). There was no recurrence of seizures. IIeus resolved on day 2 of admission, after which enteral nutrition was commenced and built up. The child had pain in both lower limbs, for which tramadol hydrochloride (1mg/kg) was given whenever required. This was the last of the symptoms to recover and continued for 8 days. The child has been asymptomatic over a follow up period of 9 months.
Case 2
A 10-year old girl presented to the emergency with a 2-day history of severe periumblical and non–colicky abdominal pain. There was neither associated abdominal distension nor vomiting. On examination the child was conscious, well oriented, with a pulse rate of 146/min and BP of 160/110 mm Hg. Sexual maturity rating was pre–pubertal and abdominal examination was unremarkable. On reviewing the history, it was found that the child had suffered recurrent episodes of pain in the past 3 years, once every 4-5 months. She had been extensively evaluated and all investigations were normal. She had received empirical anti–tubercular therapy without any response.
The child also had an episode of unexplained seizures a year prior to coming to our hospital. CSF examination and MRI brain done at that time were normal. The child was on valproate since then. Initial laboratory investigations revealed hyponatremia (serum sodium 113 meq/L). Ultrasonography and
X–ray abdomen were normal. The possibility of AIP was noted and a urinary porphobilinogen was done which was positive.
The child was managed with 10% dextrose infusion. Atenolol (1 mg/kg/day) and enalapril (0.5 mg/kg/day) were used to control blood pressure. The child showed gradual recovery over 72 hours (Table 1) with subsidence of abdominal pain. Anti–hypertensive medication was tapered and stopped. Sodium valproate was stopped and gabapentin was started at admission.
On day 4 of admission, the child developed a limp. Neurological evaluation showed a weakness (grade 2/5) in the proximal right lower limb. Sensations were preserved. A nerve conduction study suggested motor neuropathy. Physiotherapy was started. The child has now been on regular follow-up for 7 months and apart from a mild residual weakness (grade 4/5) is doing well.
Neither of the cases had a family history of porphyria. At discharge both families were educated about the disorder and its possible precipitating factors. A list of drugs that can be safely administered in porphyria was given.
Discussion
Acute intermittent porphyria is inherited as an autosomal dominant disorder with low penetrance resulting from mutation in the gene encoding the enzyme, porphobilinogendeaminase. AIP is rare and may remain undiagnosed throughout life unless an appropriate biochemical analysis is performed.90% remain biochemically and clinically normal and need a second hit, Such as exposure to certain endogenous or exogenous factors (prolonged fasting, drugs, psychosocial stress and inter current illnesses) to become symptomatic. But it is not uncommon for individuals to have acute attacks in the absence of a precipitating factor as seen in our two cases.
Symptoms are more common in women and rare before puberty. In two large series from Europe comprising a total of 269 patients, only 6 (2.2%) were less than 15 years of age at diagnosis.[1].Reproductive hormones play an important role in the clinical expression of AIP, accounting for its occurrence only after puberty.[2]
Severe abdominal pain, generalized or localized, is the most common symptom. It is seen in 85-95% patients and may be accompanied by back, buttock or limb pain. Absence of guarding is characteristic and helps to differentiate from a surgical abdomen. Constipation (50%), nausea and vomiting (50%), tachycardia (40%), hypertension (31%), urine discoloration (pink) (25%) and seizures (10–16%) are the common signs and symptoms.[3] Hyponatremia occurring as a result of a syndrome of inappropriate ant diuretic hormone secretion is well recognized in AIP patients.
AIP is known to present as a neurological emergency in the form of seizures, peripheral neuropathy and loss of consciousness. Subsequent use of common enzyme inducing anti– epileptic drugs can cause further worsening of this condition as was seen in our patients who were prescribed phenytoin and valproate respectively to control seizures at another center. Gabapentin, vigabatrin and levatiracetam have been found safe in AIP.
It is important to recognize this disorder as untreated severe acute attacks may have fatal consequences. Hematin (hemearginate) the drug of choice is not available in India. In its absence a high carbohydrate diet, with a minimum daily intake of300 g of carbohydrates is required during attacks.
Liver transplantation is curative and is indicated when there are repeated life-threatening acute attacks. Hepatocellular carcinoma and chronic renal insufficiency are important causes of morbidity and mortality on long-term follow-up. Barclay et al[4] in 1974 reported AIP in an 11-year old child who had presented with abdominal pain and generalized convulsions, and had hypertension and hyponatremia similar to our child. However, since then only few such cases have been reported in world literature. Recurrent abdominal pain, as seen here, as the sole presentation, has rarely been reported.[5]
Rarity in the pre–pubertal age group means that one would need a high index of suspicion to clinically diagnose this condition in young children. Urine for porphobilinogen detected by Watson–Schwartz test is a simple and inexpensive test. In the absence of molecular genetic testing and erythrocyte enzyme levels, it is the most readily available test. This test should be done in children with unexplained and severe acute or recurrent abdominal pain accompanied by any one of the manifestations of muscle weakness, mental changes (insomnia, hallucinations, confusion or altered consciousness), seizures, hypertension, hyponatremia, discoloration of urine on standing and family history of AIP or unexplained seizures.
References
- Goldberg A. Acute intermittent porphyria: a study of 50 cases. Q J Med.1959;28:183–209.
- Andersson C, Innala E, Bäckström T. Acute intermittent porphyria in women: clinical expression, use and experience of exogenous sex hormones. A population-based study in northern Sweden. J Intern Med. 2003;254:176–83.
- Foran SE, Abel G, Guide to the porphyrias. A historical and clinical perspective. Am J Clin Pathol. 2003;119:S86–93.
- Barclay N.Acute intermittent porphyria in childhood. A neglected diagnosis? Arch Dis Child. 1974;49:404–6.
- Rosland JH. Recurrent abdominal pain caused by acute intermittent porphyria. Tidsskr Nor Lægeforen. 2001;121:2818–20.
|
|
|
|
|
|