|
|
|
|
 |
|
| |
 |
|
|
Original Articles |
|
|
|
|
|
Keywords :
QT interval, acute variceal bleeding, bleed-precipitated hepatorenal syndrome, serum sodium, beta-blocker |
|
|
|
George Peter, Paul Cheruvathoor George, Mashhood Padincharepurathu Villyoth, Sijil Sivaraman, Rooby Erachamveettil Hamza, Suthanu Bahuleyan, Kadavanoor Srijith, Abhilash Haridas, Shanid Abdul Sathar, Srijaya Sreesh, Premalatha Narayanan, Kattoor Ramakrishnan Vinayakumar
Department of Medical
Gastroenterology,
Government Medical College,
Thiruvananthapuram, Kerala, India
Corresponding Author:
George Peter
Email: georgepeter23@gmail.com
DOI:
http://dx.doi.org/10.7869/tg.203
Abstract
Background: This study aimed to assess whether QT interval prolongation is an independent risk factor for development of hepatorenal syndrome (HRS) in cirrhotic patients with acute variceal bleeding.
Methods: 78 consecutive cirrhotic patients with acute variceal bleeding were included in the study. All patients were evaluated before bleeding (T0), during bleeding (T1) and 6 weeks later (T2).
Results: HRS developed in 14 (17.9%) patients. QT corrected by heart rate (QTc) prolonged at T1, returning towards baseline at T2 (mean ± SD; from 424.0±10.2 to 461.2±17.6 to 426.1±8.8ms, P<0.001). At T1, patients who developed HRS had longer QTc (P=0.017) and lower serum sodium (P=0.039). QTc and serum sodium independently predicted HRS; the best cut-off values were QTc > 468 ms and sodium <120 mEq/L. Patients on beta-blocker were found to have significant risk for developing HRS (p=0.040). Based on these three factors, the risk for HRS was nil for patients without risk factors; 6.1%, 11.1%, and 83.3% for those with one, two or three risk factors, respectively (p<0.001).
Conclusions: Acute variceal bleeding causes further prolongation of QTc in cirrhosis. The combination of beta-blocker, QTc interval and serum sodium can aid in early detection of patients at increased risk of developing bleed-precipitated HRS, thus improving their outcome.
|
48uep6bbphidvals|627 48uep6bbph|2000F98CTab_Articles|Fulltext Cardiac dysfunction in cirrhotic patients characterized by impaired contractile responsiveness to stress and/or deranged diastolic relaxation with electro-physiological alterations, after exclusion of known cardiac diseases, is termed cirrhotic cardiomyopathy.[1] The characteristic finding in electrocardiogram (ECG) is prolongation of QT interval,[2] which may be seen in approximately half of the patients.[3] Though the exact pathophysiology of this entity is unclear, sympathetic nervous system activation has been widely regarded as a contributing factor, as evidenced by correlation between plasma norepinephrine concentration and QT interval.[4] Role of abnormal membrane biochemical function, over-activity of cardiac depressants like nitric oxide, fibrosis and edema of cardiac muscle, have also been implicated in its pathophysiology.[5] Studies have demonstrated normalization of QT interval with chronic â blockade in those with prolonged QT interval at base line.[6]
Whenever the cirrhotic heart is subjected to stressful events like acute gastrointestinal bleeding, sepsis, liver transplantation, insertion of transjugular intrahepatic portosystemic stent– shunts (TIPS), the covert heart failure becomes overt worsening morbidity.[7,8] These conditions cause further prolongation of the QT interval and it’s prognostic significance for acute gastrointestinal bleeding has been examined recently. Prolonged QT interval has been found to directly correlate with mortality.[9]
Acute variceal bleeding is a well-recognized precipitant for hepatorenal syndrome (HRS), especially in advanced cirrhosis.[10] Reduced cardiac contractility due to cirrhotic cardiomyopathy, along with reduced cardiac output and mean arterial pressure, predisposes to renal dysfunction and HRS.[11] Hence, whether QT interval prolongation, a well-accepted marker of cardiac dysfunction in cirrhosis, can predict the development of HRS following an episode of acute variceal bleeding remain uncertain. The aim of this study was to assess whether QT interval prolongation is an independent risk factor for development of HRS in cirrhotic patients with acute variceal bleeding.
Methods
This prospective observational study was conducted at a tertiary care centre in South India, from November 2012 to November 2013. Study subjects were selected from consecutive cirrhotic patients admitted with acute variceal bleeding. The diagnosis of cirrhosis was based on a combination of clinical, biochemical, imaging and endoscopic evidence of esophageal or gastric varices, and histological features, where available.
Only those patients who had been evaluated within 6 months prior to bleed, including a good quality ECG tracing, were included in the study. Exclusion criteria were age less than 18 years, diabetes mellitus, known cardiac illness including conduction abnormalities, family history of sudden cardiac death, electrolyte disturbances (hypocalcemia, hypomagnesemia, hypokalemia), patients on drugs affecting QT interval except â blockers and quinolones.
Data was recorded at three time points. Data recorded prior to variceal bleed was taken as baseline (T0). Patients underwent thorough clinical evaluation, lab investigations and a 12-lead ECG recording, at the time of variceal bleed (T1), and 6 weeks later (T2). The diagnosis of acute variceal bleeding was confirmed with upper gastrointestinal endoscopy. ECG recording was obtained using a Cardioline Delta 3 Plus ECG Unit at a paper speed of 25 mm/s. The QT interval length was measured manually from the beginning of the Q wave (or R wave) to the end of the T wave in all 12 leads. T wave endpoints were determined by drawing the base line and a line tangent to the T wave. Measurements were taken from leads in which the end of the T wave could be clearly delineated. Two consecutive QT intervals were measured in each lead, and the arithmetic mean was calculated. All ECG measurements were made by two independent observers blinded to the patient data. Correction for heart rate (QTc) was made based on the ‘cirrhosis-specific’ formula (QTc = QT/RR1/3.02).[12] The diagnosis of HRS was based on the revised International Ascites Club criteria for the diagnosis of HRS.[13-15] The criteria includes: (1) cirrhosis with ascites; (2) serum creatinine >1.5 mg/dl; (3) no improvement in serum creatinine (decrease to a level <1.5 mg/ dl) after two days of diuretics withdrawal and volume expansion with albumin (1g/kg of body weight up to a maximum of 100 g/ day); (4) absence of shock; (5) no current or recent treatment with nephrotoxic drugs; and (6) absence of parenchymal kidney disease, as indicated by a urinary protein concentration <500 mg/day, a urinary red blood cell count <50 cells per high power
field, and normal renal ultrasonography. Variables were expressed as mean ± SD or median/ interquartile range, frequency and percentage, as appropriate. Differences between and within groups were assessed by unpaired or paired Student t-tests, Mann–Whitney U or Wilcoxon tests, and Chi-square test or ANOVA (analysis of variance) for repeated measures, as appropriate. Spearman’s and Pearson’s tests were used to determine correlations, as appropriate. Binary logistic regression was performed for those variables which were found to be significant during the univariate analysis. Receiver operating characteristic (ROC) curve analysis was applied to the independent prognostic factors, to calculate the area under the curve (AUROC), 95% confidence interval (CI) and the ideal cut-off value. Statistical analysis was performed using SPSS v17.0. A p-value <0.05 was considered statistically significant.
Results
Of the 78 patients included in the study, 66 (84.6%) were males and 12 (15.4%) were females. The mean age was 50.76±12.24 years and 53.83±9.55 years in males and females, respectively. The etiology of cirrhosis was alcohol in 42 patients (53.8%), hepatitis B virus (HBV) in 17 (21.8%), alcohol + HBV in 7 (8.9%), hepatitis C virus (HCV) in 9 (11.5%), autoimmune hepatitis in 2 (2.6%) and alcohol + HCV in 1 (1.2%). 44 patients (56.4%) were on â blocker (propranolol) therapy and 34 patients (43.6%) were either not compliant to therapy or were not on â blocker prophylaxis. Hemorrhage ensued from esophageal varices in 74 patients (94.8%) and combined esophageal and gastric varices in 4 patients (5.2%). Presence of portal vein thrombosis by ultrasound Doppler was noted in 10 patients.
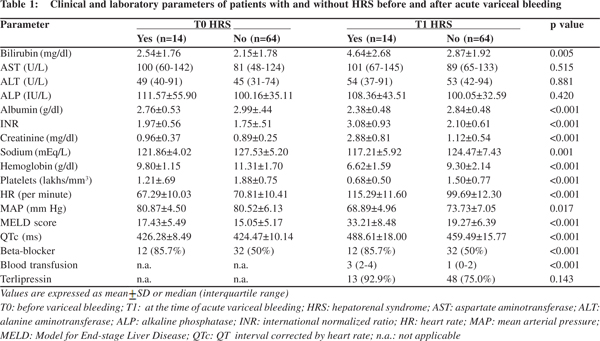
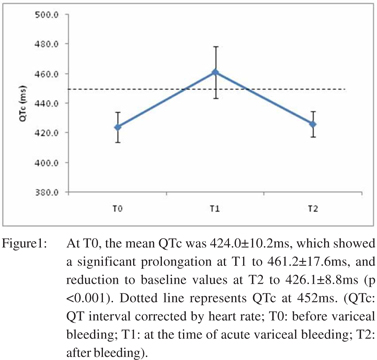
Among the 78 patients, 28 (35.9%) developed acute kidney injury (AKI), of which 14 (17.9%) developed HRS. Table 1 represents the comparison of clinical and laboratory parameters of patients who developed HRS, and those who did not, at baseline (T0) and at the time of variceal bleeding (T1). The mean duration between T0 and T1 was 60.6 ± 43.9 days. Prolonged QTc was present in 53 patients (67.9%) at T1. Etiology of cirrhosis did not have a significant correlation with prolonged QTc. Initiation of terlipressin did not show significant association with QTc prolongation at T1. At T0, the mean QTc was 424.0 ± 10.2 ms, which showed a significant prolongation at T1 to 461.2 ± 17.6 ms, and reduction to baseline values at T2 (in survivors) to 426.1 ± 8.8ms (p <0.001) (Figure 1). QTc showed significant prolongation from baseline in those who developed HRS (488.61 ± 17.99 ms vs. 459.49 ± 15.76 ms, p <0.001) (Figure 2). The degree of QT prolongation measured as DT0-T1QTc was significantly higher in the group which developed HRS than those who did not. (62.3 ± 12.3 ms vs. 35.0 ± 13.7 ms; p <0.001).
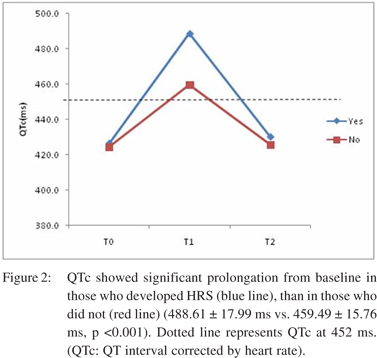
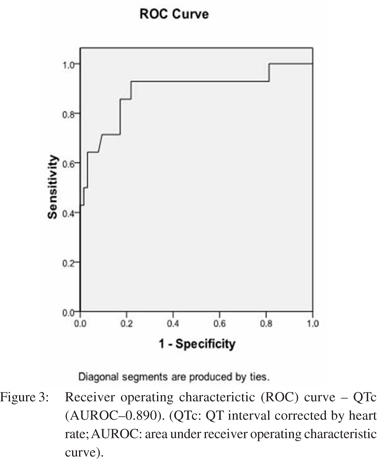
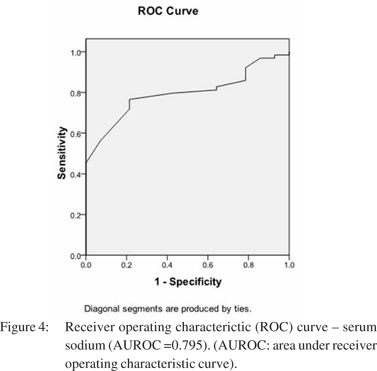
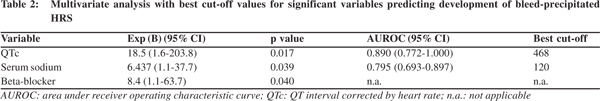
 â blocker therapy did not hinder bleed induced QT prolongation. DT0-T1 QTc in those on propranolol was 41.6 ± 19.4 ms, whereas it was 37.8 ± 13.4 ms for those who were not on it (p=0.33). Of the 64 patients who did not develop HRS, 50% were on beta blockers, whereas 85.7% (12 ) of patients who developed HRS were on â blocker therapy (p <0.001). None of the patients showed any significant ST/T changes in ECG suggestive of myocardial ischemia, in any of the three ECG recordings. Mean arterial pressure and hemoglobin showed a significant drop from T0 to T1, with return to baseline values in survivors, at T2. Heart rate also recorded a significant rise from T0 to T1, which approached baseline values at T2, in survivors. The MELD score had a significant rise from T0 to T1, before returning to pre-bleed levels at T2 in survivors (p <0.001). At T1, the hemodynamic parameters and MELD score showed direct correlation with DT0-T1QTc. DT0-T1 heart rate and DT0-T1 MELD score directly correlated with DT0-T1QTc (R = 0.562 and 0.587 respectively, p <0.001), while DT0-T1 hemoglobin and DT0-T1 mean arterial pressure had inverse correlation with DT0- T1QTc (R = -0.418 and -0.478 respectively, p <0.001). Overall 6- week mortality rate was 16.7% (13 patients). HRS was documented in six out of these 13 patients (46.2%, p=0.012). On univariate analysis, hemoglobin, heart rate, mean arterial pressure, serum sodium, platelet count, MELD score, QTc interval at T1, use of â blocker, and number of blood transfusions were associated with development of HRS. On binary logistic regression, QTc interval at T1, serum sodium and use of beta blocker emerged as independent predictors for development of bleed precipitated HRS. ROC curves were plotted for these three variables (Figure 3, 4). Table 2 represents the results of multivariate analysis with best cut-off values for the significant variables. The cumulative risk for development of HRS was calculated based on these three independent predictive factors with their respective cut-off values. Of the 15 patients with no risk factor, none of them developed HRS. Meanwhile, 2/33 (6.1%) patients with 1 risk factor, 2/18(11.1%) patients with 2 risk factors and 10/12 (83.3%) with all three risk factors developed HRS (p <0.001).
â blocker therapy did not hinder bleed induced QT prolongation. DT0-T1 QTc in those on propranolol was 41.6 ± 19.4 ms, whereas it was 37.8 ± 13.4 ms for those who were not on it (p=0.33). Of the 64 patients who did not develop HRS, 50% were on beta blockers, whereas 85.7% (12 ) of patients who developed HRS were on â blocker therapy (p <0.001). None of the patients showed any significant ST/T changes in ECG suggestive of myocardial ischemia, in any of the three ECG recordings. Mean arterial pressure and hemoglobin showed a significant drop from T0 to T1, with return to baseline values in survivors, at T2. Heart rate also recorded a significant rise from T0 to T1, which approached baseline values at T2, in survivors. The MELD score had a significant rise from T0 to T1, before returning to pre-bleed levels at T2 in survivors (p <0.001). At T1, the hemodynamic parameters and MELD score showed direct correlation with DT0-T1QTc. DT0-T1 heart rate and DT0-T1 MELD score directly correlated with DT0-T1QTc (R = 0.562 and 0.587 respectively, p <0.001), while DT0-T1 hemoglobin and DT0-T1 mean arterial pressure had inverse correlation with DT0- T1QTc (R = -0.418 and -0.478 respectively, p <0.001). Overall 6- week mortality rate was 16.7% (13 patients). HRS was documented in six out of these 13 patients (46.2%, p=0.012). On univariate analysis, hemoglobin, heart rate, mean arterial pressure, serum sodium, platelet count, MELD score, QTc interval at T1, use of â blocker, and number of blood transfusions were associated with development of HRS. On binary logistic regression, QTc interval at T1, serum sodium and use of beta blocker emerged as independent predictors for development of bleed precipitated HRS. ROC curves were plotted for these three variables (Figure 3, 4). Table 2 represents the results of multivariate analysis with best cut-off values for the significant variables. The cumulative risk for development of HRS was calculated based on these three independent predictive factors with their respective cut-off values. Of the 15 patients with no risk factor, none of them developed HRS. Meanwhile, 2/33 (6.1%) patients with 1 risk factor, 2/18(11.1%) patients with 2 risk factors and 10/12 (83.3%) with all three risk factors developed HRS (p <0.001).
 Discussion
This study confirms the occurrence of further prolongation of QT interval accompanying an episode of acute variceal bleeding, which is independent of the etiology of underlying cirrhosis. It was established that the further prolongation of QT interval from baseline pre-bleeding values, was a result of the acute insult itself, as evident by the return of QT interval to near baseline values 6 weeks after bleeding; even in the group which developed HRS. The beneficial effect of â blockers in cirrhotic patients which works by shortening the QT interval prior to liver transplantation, by improving vagal cardiac modulation has evoked interest.[16,17] Chronic â blockade although effective in shortening the prolonged QT interval at baseline,6 was not able to prevent the bleed induced QT prolongation in our study group. To the best of our knowledge the prognostic significance of DQTc in stressful conditions has never been evaluated.
DQTc had direct correlation with various hemodynamic parameters and MELD scores, demonstrating the importance of QT interval prolongation as a marker of cirrhotic cardiomyopathy. Along with prolonged QTc, DQTc also emerged as a reliable predictor of development of bleed precipitated HRS. However, the availability of a pre-bleed ECG significantly hinders the clinical utility of DQTc. Even though the exact mechanism of prolonged QTc in cirrhosis remains obscure, there is sufficient data to prove normalization of QT interval following liver transplantation,[18,19] suggesting that QT prolongation is directly correlated with degree of liver dysfunction. Various commonly used prognostic indicators in cirrhosis, including MELD scores, do not incorporate the role of cardiac dysfunction in development of common complications including HRS. QTc could be a strong contender in this regard, especially in the wake of recent studies supporting its role in clinical practice.[20-23] There is ample evidence of cardiac conduction abnormalities in cirrhosis,[2,24,25] which predispose to acute cardiac events, especially in the setting of a gastrointestinal bleed. However, this entity is underestimated, probably due to the restricted use of continuous ECG monitoring in such patients.
In our study, three variables including QTc, serum sodium and use of beta-blocker emerged as independent risk factors for development of bleed-precipitated HRS. Apart from serving as an easily available, non-invasive clinical tool for predicting HRS in the context of variceal bleeding, QTc prolongation has established the role of cardiac dysfunction in the pathophysiology of HRS. Lower values of serum sodium have been identified with increased risk for development of HRS. Our study confirms its predictive role in bleed-precipitated HRS. A recent study has suggested the role of serum sodium as an independent predictor of short term mortality in patients with HRS, and also in selecting patients requiring early liver transplantation following onset of HRS.[26] Recently there has been considerable interest in the changing role of â blockers in advanced cirrhosis, in view of its negative impact on cardiac reserve, resulting in reduced perfusion to kidneys during periods of stress, and precipitation of hepatorenal syndrome.[27-29] Our study supports these findings of deleterious effect of this drug in bleed-precipitated HRS. A recent study contraindicates â blockers in cirrhotic patients with refractory ascites.[27]
Discussion
This study confirms the occurrence of further prolongation of QT interval accompanying an episode of acute variceal bleeding, which is independent of the etiology of underlying cirrhosis. It was established that the further prolongation of QT interval from baseline pre-bleeding values, was a result of the acute insult itself, as evident by the return of QT interval to near baseline values 6 weeks after bleeding; even in the group which developed HRS. The beneficial effect of â blockers in cirrhotic patients which works by shortening the QT interval prior to liver transplantation, by improving vagal cardiac modulation has evoked interest.[16,17] Chronic â blockade although effective in shortening the prolonged QT interval at baseline,6 was not able to prevent the bleed induced QT prolongation in our study group. To the best of our knowledge the prognostic significance of DQTc in stressful conditions has never been evaluated.
DQTc had direct correlation with various hemodynamic parameters and MELD scores, demonstrating the importance of QT interval prolongation as a marker of cirrhotic cardiomyopathy. Along with prolonged QTc, DQTc also emerged as a reliable predictor of development of bleed precipitated HRS. However, the availability of a pre-bleed ECG significantly hinders the clinical utility of DQTc. Even though the exact mechanism of prolonged QTc in cirrhosis remains obscure, there is sufficient data to prove normalization of QT interval following liver transplantation,[18,19] suggesting that QT prolongation is directly correlated with degree of liver dysfunction. Various commonly used prognostic indicators in cirrhosis, including MELD scores, do not incorporate the role of cardiac dysfunction in development of common complications including HRS. QTc could be a strong contender in this regard, especially in the wake of recent studies supporting its role in clinical practice.[20-23] There is ample evidence of cardiac conduction abnormalities in cirrhosis,[2,24,25] which predispose to acute cardiac events, especially in the setting of a gastrointestinal bleed. However, this entity is underestimated, probably due to the restricted use of continuous ECG monitoring in such patients.
In our study, three variables including QTc, serum sodium and use of beta-blocker emerged as independent risk factors for development of bleed-precipitated HRS. Apart from serving as an easily available, non-invasive clinical tool for predicting HRS in the context of variceal bleeding, QTc prolongation has established the role of cardiac dysfunction in the pathophysiology of HRS. Lower values of serum sodium have been identified with increased risk for development of HRS. Our study confirms its predictive role in bleed-precipitated HRS. A recent study has suggested the role of serum sodium as an independent predictor of short term mortality in patients with HRS, and also in selecting patients requiring early liver transplantation following onset of HRS.[26] Recently there has been considerable interest in the changing role of â blockers in advanced cirrhosis, in view of its negative impact on cardiac reserve, resulting in reduced perfusion to kidneys during periods of stress, and precipitation of hepatorenal syndrome.[27-29] Our study supports these findings of deleterious effect of this drug in bleed-precipitated HRS. A recent study contraindicates â blockers in cirrhotic patients with refractory ascites.[27]
One limitation of our study was the need for a pre-bleed ECG for documentation of DQTc, which has inadvertently led to inclusion of higher number of patients with advanced cirrhosis (Child-Pugh C patients), as they are more likely to possess a pre-bleed ECG recorded during their previous hospitalizations or hospital visits for an unrelated complication. This might have contributed to the higher incidence of HRS in our study population. The use of quinolones, a drug group known to produce prolongation of ventricular re-polarization, was permitted in our study group. Norfloxacin was permitted in this study, but is not known to cause significant QT prolongation, unlike moxifloxacin and ciprofloxacin.30 Lastly the co-existence of other complications like spontaneous bacterial peritonitis, alcoholic hepatitis and sepsis could have influenced the results. However, considering the nature of these complications, further longitudinal studies are required to assess the impact of cardiac dysfunction in the final outcome. In conclusion, our study illustrates further prolongation of QT interval from base line in cirrhotic patients, reflecting ventricular re-polarization abnormalities during acute variceal bleeding. In combination with serum sodium and use of â blockers, prolonged QTc comprises an invaluable tool for predicting HRS development in cirrhotic patients with acute variceal bleeding. This can help clinicians to triage patients at risk early and optimize their management. The use of â blocker, presence of serum sodium d”120 mEq/L, and QTc e”468 ms at the time of acute variceal bleeding can stratify risk of bleedprecipitated HRS; with risk ranging from none in patients with no risk factors to 83.3% in patients with all three risk factors. However further studies are warranted to confirm the impact of QTc prolongation in cirrhosis during stressful events.
References
- Moller S, Henriksen JH. Cirrhotic cardiomyopathy. J Hepatol. 2010;53:179–90.
- Zambruni A, Trevisani F, Caraceni P, Bernardi M. Cardiac electrophysiological abnormalities in patients with cirrhosis. J Hepatol. 2006;44:994–1002.
- Bernardi M, Calandra S, Colantoni A, Trevisani F, Raimondo ML, Sica G, et al. Q-T interval prolongation in cirrhosis: prevalence, relationship with severity, and etiology of the disease and possible pathogenetic factors. Hepatology. 1998;27:28–34.
- Bernardi M, Maggioli C, Dibra V, Zaccherini G. QT interval prolongation in liver cirrhosis: innocent bystander or serious threat? Expert Rev Gastroenterol Hepatol. 2012;6:57–66.
- Liu H, Song D, Lee SS. Cirrhotic cardiomyopathy. Gastroenterol Clin Biol. 2002;26:842–7.
- Zambruni A, Trevisani F, Di Micoli A, Savelli F, Berzigotti A,Bracci E, et al. Effect of chronic beta-blockade on QT interval in patients with liver cirrhosis. J Hepatol. 2008;48:415–21.
- Baik SK, Lee SS. Cirrhotic cardiomyopathy. Orphanet Encyclopedia, January 2005; [Available at: http://www.orpha.net/data/patho/GB/uk-CirrhoticCardiomyopathy.pdf]
- Zamirian M, Tavassoli M, Aghasadeghi K. Corrected QT interval and QT dispersion in cirrhotic patients before and after liver transplantation. Arch Iran Med. 2012;15:375–7.
- Trevisani F, Di Micoli A, Zambruni A, Biselli M, Santi V, Erroi V, et al. QT interval prolongation by acute gastrointestinal bleeding in patients with cirrhosis. Liver Int. 2012;32:1510–5.
- Angeli P, Morando F. Optimal management of hepatorenal syndrome in patients with cirrhosis. Hepat Med. 2010;2:87–98.
- Ruiz-del-Arbol L, Monescillo A, Arocena C, Valer P, Gines P, Moreira V, et al. Circulatory function and hepatorenal syndrome in cirrhosis. Hepatology. 2005;42:439–47.
- Zambruni A, Di Micoli A, Lubisco A, Domenicali M, Trevisani F, Bernardi M. QT interval correction in patients with cirrhosis. J Cardiovasc Electrophysiol. 2007;18:77–82.
- European Association for the Study of the Liver. EASL clinical practice guidelines on the management of ascites, spontaneous bacterial peritonitis, and hepatorenal syndrome in cirrhosis. J Hepatol. 2010;53:397–417.
- Salerno F, Gerbes A, Gines P, Wong F, Arroyo V. Diagnosis, prevention and treatment of hepatorenal syndrome in cirrhosis. Gut. 2007;56:1310–8.
- Salerno F, Cazzaniga M, Merli M, Spinzi G, Saibeni S, Salmi A, et al. Diagnosis, treatment and survival of patients with hepatorenal syndrome: a survey on daily medical practice. J Hepatol. 2011;55:1241–8.
- Kim YK, Hwang GS, Shin WJ, Bang JY, Cho SK, Han SM. Effect of propranolol on the relationship between QT interval and vagal modulation of heart rate variability in cirrhotic patients awaiting liver transplantation. Transplant Proc. 2011;43:1654–9.
- Mohamed R, Forsey PR, Davies MK, Neuberger JM. Effect of liver transplantation on QT interval prolongation and autonomic dysfunction in end-stage liver disease. Hepatology. 1996;23:1128–34.
- Adigun AQ, Pinto AG, Flockhart DA, Gorski JC, Li L, Hall SD, et al. Effect of cirrhosis and liver transplantation on the gender difference in QT interval. Am J Cardiol. 2005;95:691–4.
- Shin WJ, Kim YK, Song JG, Kim SH, Choi SS, Song JH, et al. Alterations in QT interval in patients undergoing living donor liver transplantation. Transplant Proc. 2011;43:170–3.
- Bernardi M, Calandra S, Colantoni A, Trevisani F, Raimondo ML, Sica G, et al. Q-T interval prolongation in cirrhosis: prevalence, relationship with severity, and etiology of the disease and possible pathogenetic factors. Hepatology. 1998;27:28–34.
- Bal JS, Thuluvath PJ. Prolongation of QTc interval: relationship with etiology and severity of liver disease, mortality and liver transplantation. Liver Int. 2003;23:243–8.
- Henriksen JH, Gotze JP, Fuglsang S, Christensen E, Bendtsen F, Moller S. Increased circulating pro-brain natriuretic peptide (proBNP) and brain natriuretic peptide (BNP) in patients with cirrhosis: relation to cardiovascular dysfunction and severity of disease. Gut. 2003;52:1511–7.
- Hansen S, Moller S, Bendtsen F, Jensen G, Henriksen JH. Diurnal variation and dispersion in QT interval in cirrhosis: relation to haemodynamic changes. J Hepatol. 2007;47:373–80.
- Wong F. Cirrhotic cardiomyopathy. Hepatol Int. 2009;3:294–304.
- Rudy Y. Molecular basis of cardiac action potential repolarization. Ann N Y Acad Sci. 2008;1123:113–8.
- Licata A, Maida M, Bonaccorso A, Macaluso FS, Cappello M, Craxi A, et al. Clinical course and prognostic factors of hepatorenal syndrome: A retrospective single-center cohort study. World J Hepatol. 2013;5:685–91.
- Serste T, Melot C, Francoz C, Durand F, Rautou PE, Valla D, et al. Deleterious effects of beta-blockers on survival in patients with cirrhosis and refractory ascites. Hepatology. 2010;52:1017–22.
- Krag A, Bendtsen F, Henriksen JH, Moller S. Low cardiac output predicts development of hepatorenal syndrome and survival in patients with cirrhosis and ascites. Gut. 2010;59:105–10.
- Serste T, Francoz C, Durand F, Rautou PE, Melot C, Valla D, et al. Beta-blockers cause paracentesis-induced circulatory dysfunction in patients with cirrhosis and refractory ascites: a cross-over study. J Hepatol. 2011;55:794–9.
- Falagas ME, Rafailidis PI, Rosmarakis ES. Arrhythmias associated with fluoroquinolone therapy. Int J Antimicrob Agents. 2007;29:374–9.
|
|
|
|
|
|