|
|
|
|
 |
|
| |
 |
|
|
Original Articles |
|
|
|
|
|
Keywords :
Cirrhosis, hepatic vena cava disease, Budd-Chiari syndrome, hepatic venous outflow obstruction, bacterial infection. |
|
|
Santosh Man Shrestha, Shobhana Shrestha
Liver Foundation Nepal,
Sitapaela Height, Kathmandu, Nepal
Corresponding Author:
Dr. Santosh Man Shrestha
Email: smshrestha1938@gmail.com
DOI:
http://dx.doi.org/10.7869/tg.186
Abstract
Background: Disease of hepatic portion of the inferior vena cava (IVC) now renamed hepatic vena cava syndrome (HVCS) is an insidious onset chronic disease characterized by recurrent acute exacerbations and development of cavo-caval collaterals. In adults it is complicated by high incidence of liver cirrhosis. Aim of this study was to assess the incidence of liver cirrhosis in children with HVCS and discuss its pathogenesis.
Method: One hundred and seventy eight children with HVCS were followed up with ultrasonography (USG), routine hematology and liver tests. During acute exacerbations, cultures of blood and ascitic fluid for aerobic microorganisms were also done. Diagnosis of liver cirrhosis was based on transformation of the liver parenchymal echo-texture from normal to uniformly coarse with rounded edges in follow-up USG. It was confirmed by direct inspection of the liver or by biopsy in six. The duration from disease onset to detection of liver cirrhosis was also assessed.
Results: HVCS was seen in children from poor socio-economic background. Forty nine patients (27.5%) developed liver cirrhosis within a few months to a few years of onset of the disease. Development of cirrhosis was related to frequency or severity of acute exacerbations and not to duration of illness or severity of the caval lesion.
Conclusion: The cause of cirrhosis in HVCS was the ischemic injury of the hepatocytes and sinusoids following thrombosis of the IVC and/or intra-hepatic veins during severe acute episodes or exacerbations precipitated by bacterial infection. Since such acute exacerbations are amenable to medical treatment, HVCS may be a cause of preventable liver cirrhosis in developing countries.
|
48uep6bbphidvals|645 48uep6bbph|2000F98CTab_Articles|Fulltext Cirrhosis is the end stage of many forms of liver injury. Major causes of cirrhosis in the world are chronic infections with hepatitis B and hepatitis C virus, and chronic alcohol abuse. It takes about 20 to 25 years of exposure to these agents for liver cirrhosis to develop.[1] The causes of liver cirrhosis in children are slightly different. In children cirrhosis is related to inborn errors of metabolism like Wilson’s disease or biliary atresia and autoimmune hepatitis.[2,3] Some unique types of cirrhosis in children, specific to certain geographic regions, had been described in the past, such as veno-occlusive disease (VOD) - now renamed sinusoidal obstruction syndrome (SOS) from Jamaica in 1950s-1960s[4] and Indian childhood cirrhosis from India during 1950s to 1990s.[5,6] We report here liver cirrhosis in children due to hepatic vena cava syndrome (HVCS), a disease of the hepatic portion of the IVC, endemic in Nepal.[7]
HVCS was reported from Asian and African countries[7-15] under various diagnoses such as Budd Chiari Syndrome (BCS), membranous obstruction of the IVC (MOVC), IVC obstruction or coarctation of inferior vena cava. In the past it was also reported from the West as hepatic vein occlusion.[16-18] It is a chronic disease that begins insidiously, has a long asymptomatic period and clinically manifests with recurrent acute exacerbations. Recurrent exacerbations clinically manifest as recurrent jaundice, biochemically as recurrent mild elevation of serum bilirubin and or alanine aminotransferase (ALT), and sonographically as deposition of layers of thrombus on posterior wall of the IVC at or close to the initial lesion. Severe acute exacerbations are characterized by fever, jaundice (related to bacterial infection), hepatomegaly, ascites, pleural effusion or transient portal hypertension (related to hepatic venous outflow obstruction) or edema of the legs (caval obstruction). Other clinical features include intermittent upper abdomen discomfort usually after food or exertion. The disease is sometime diagnosed following fortuitous detection of hepatomegaly or splenomegaly or dilated superficial veins in
the body trunk. Liver function tests are normal or show minimal to mild elevation of bilirubin or ALT. During acute exacerbations both are elevated. Ascites and pleural effusion may occur during acute exacerbations. Ascites has high protein content and a high serum-ascitic albumin gradient showed evidence of bacterial peritonitis.[19] Pleural effusion is usually minimal, but rarely it may be severe and recurrent.[20]
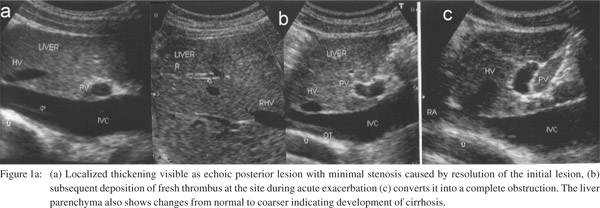
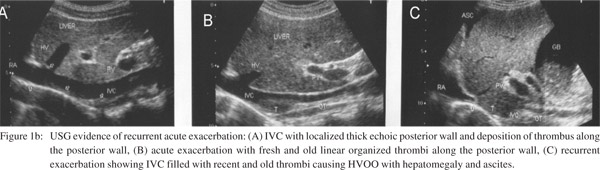
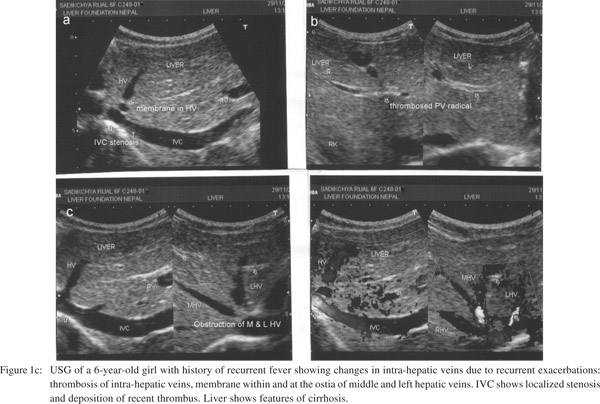
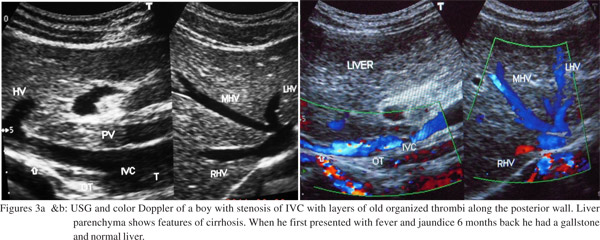
Liver biopsy or cavogram are used to confirm the diagnosis in suspected patients. Acute centrilobular congestion and atrophic hepatic cords seen during acute exacerbations are diagnostic. Biopsy findings however vary depending on the phase of the disease from normal or minimal changes like sinusoidal dilatation with mild central vein fibrosis or obstruction; periportal fibrosis or inflammatory infiltration; to endophlebitis of sublobular vein, or diffuse endophlebitis of intra-hepatic veins, lobular fibrosis or true cirrhosis.[21] A cavogram may miss the diagnosis in patients with minimal stenosis or in advanced disease when the contrast medium rapidly runs off into large collaterals and fails to outline the caval obstruction.[14] USG is specific and sensitive for the detection of HVCS and should be preferred as first line of diagnosis.[21-23] It provides information about the stage of IVC lesion (Figure 1a), evidence of acute exacerbation in the form of thrombi of different ages along the posterior wall of the IVC (Figure 1b), changes in intra-hepatic veins (Figure 1c), presence of ascites, and the state of liver parenchyma.
Diagnosis of this disease thus depends on the awareness of the condition and the use of USG. The disease had been prevalent in the West[16-18,24-28] and in Japan.[8-10] Now it is confined to the developing world. It has been reported from China,29 Taiwan,30 Korea,[31] India,[11-13] Nepal,[7] South Africa,[14,15] Egypt,[32] and from Europe in patients of North African origin.[21] Its prevalence in different geographic areas of the world has been inversely related to the standard of hygiene.[33] Liver cirrhosis is a common complication of this disease in adults (Table 1). The disease has been recorded in children[17,21,28,32,34,35] and liver cirrhosis develops as a complication in them.[17,21,32] This study aimed to assess the incidence of liver cirrhosis in children with HVCS and discuss its pathogenesis.
 Methods
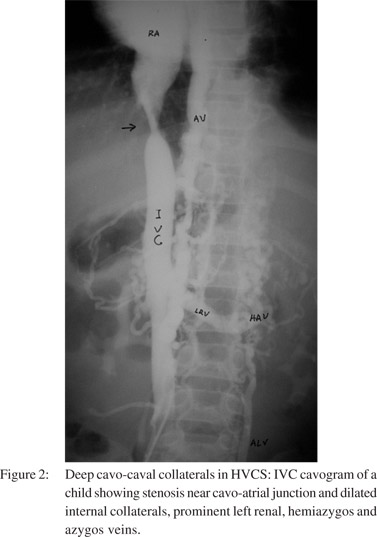
This study was based on 178 children (105 males and 73 females) with HVCS seen from 1997 to 2012. Patients with HVCS came from different parts of Nepal, but all belonged to poor socioeconomic background. Diagnosis of HVCS was based on i) USG finding of the localized lesion in the IVC opposite the site of entry of hepatic veins and ii) evidence of recurrent acute exacerbations iii) cavo-caval collaterals. It was confirmed by an inferior vena cavogram (Figure 2) in six children and by liver biopsy in five. Patients were followed at monthly intervals for first six months and then yearly with USG and serum bilirubin and ALT monitoring. Patient experiencing acute exacerbations were also tested for total and differential white blood cell counts and liver function tests. Febrile patients not started on antibiotics underwent blood cultures for aerobic microorganisms. Patients with significant ascites underwent diagnostic paracentesis with culture for aerobic microorganisms and routine tests including serum protein, albumin, serum ascites-albumin gradient (SAAG), total and differential white blood cell and absolute neutrophil counts.[36] Serological tests for hepatitis B (HBsAg) and hepatitis C (anti- HCV) were carried out for all patients. Patients found HBsAg positive were further tested for HBeAg, anti-HBe and HBVDNA levels. Those presenting with acute ALT elevation five times above normal, were tested for serological markers of acute hepatitis A (anti-HAV IgM), hepatitis B (anti-HBc IgM), and hepatitis E (anti-HEV IgM).
Methods
This study was based on 178 children (105 males and 73 females) with HVCS seen from 1997 to 2012. Patients with HVCS came from different parts of Nepal, but all belonged to poor socioeconomic background. Diagnosis of HVCS was based on i) USG finding of the localized lesion in the IVC opposite the site of entry of hepatic veins and ii) evidence of recurrent acute exacerbations iii) cavo-caval collaterals. It was confirmed by an inferior vena cavogram (Figure 2) in six children and by liver biopsy in five. Patients were followed at monthly intervals for first six months and then yearly with USG and serum bilirubin and ALT monitoring. Patient experiencing acute exacerbations were also tested for total and differential white blood cell counts and liver function tests. Febrile patients not started on antibiotics underwent blood cultures for aerobic microorganisms. Patients with significant ascites underwent diagnostic paracentesis with culture for aerobic microorganisms and routine tests including serum protein, albumin, serum ascites-albumin gradient (SAAG), total and differential white blood cell and absolute neutrophil counts.[36] Serological tests for hepatitis B (HBsAg) and hepatitis C (anti- HCV) were carried out for all patients. Patients found HBsAg positive were further tested for HBeAg, anti-HBe and HBVDNA levels. Those presenting with acute ALT elevation five times above normal, were tested for serological markers of acute hepatitis A (anti-HAV IgM), hepatitis B (anti-HBc IgM), and hepatitis E (anti-HEV IgM).
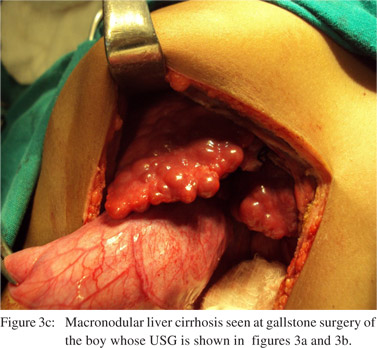
The diagnosis of cirrhosis was based on transformation of liver USG echo-texture from normal to uniformly coarse with round edges and irregular surface (Figures 3a & 3b). This assumption was based on our previous study of the disease in adults with USG and liver biopsy.[23] Development of liver cirrhosis was confirmed in this study by liver biopsy in three and by direct inspection of the liver surface (Figure 3c) in another three children (one each at laparotomy and laparoscopy for removal of gall stones and one at autopsy). The duration of detection of liver cirrhosis from onset of illness was also assessed. Since Wilson’s disease is an important cause of liver cirrhosis in children, all children diagnosed with cirrhosis underwent slit-lamp examination of their cornea for Kayser- Fleischer rings. Detailed copper studies, including estimation of serum and 24-hour urinary copper and ceruloplasmin levels were done in 20 patients only. Liver biopsy of a child was also subjected to rhodanine stain for copper. No surgical or radiological procedures were carried out to correct the caval lesions. Patients with acute exacerbations were treated with high dose oral antibiotic either ciprofloxacin or cefixime for 8 weeks with diuretics and nutritional supplements where necessary.
 Results
The majority (90%) of patients initially presented with fever and jaundice. They also had a puffy face, edema of legs and ascites in some patients and bleeding from esophageal varices in a few (Table 2). Others were detected because of intermittent upper abdominal discomfort or pain, recurrent infection at other sites with ALT elevation and mild hepatomegaly or splenomegaly. The USG showed minimal to mild stenosis of the IVC at the cavo-atrial junction with localized recent and old thrombi on the posterior wall. Forty patients (22%) gave history of recurrent fever, diarrhea or pneumonia in the past.
Results
The majority (90%) of patients initially presented with fever and jaundice. They also had a puffy face, edema of legs and ascites in some patients and bleeding from esophageal varices in a few (Table 2). Others were detected because of intermittent upper abdominal discomfort or pain, recurrent infection at other sites with ALT elevation and mild hepatomegaly or splenomegaly. The USG showed minimal to mild stenosis of the IVC at the cavo-atrial junction with localized recent and old thrombi on the posterior wall. Forty patients (22%) gave history of recurrent fever, diarrhea or pneumonia in the past.
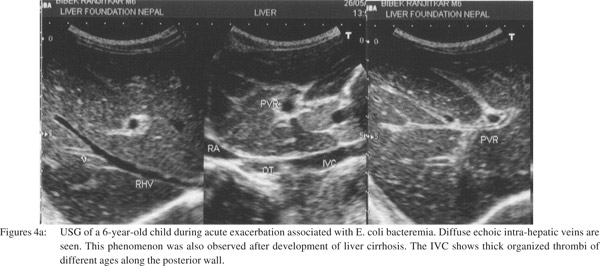
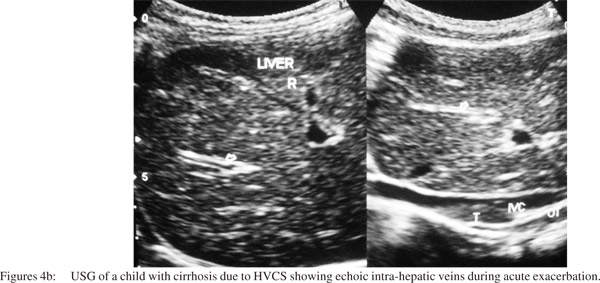 Aerobic blood culture was done in 71 patients who initially presented with fever and had not received antibiotics. Bacteremia was detected in 43 (60.5%) patients. Gram negative organisms were detected in 40, mainly Escherichia coli and Klebsiella species. Staphylococcus aureus was detected in three cases. Ascitic fluid was examined in five patients. It showed high protein, high serum-ascitic fluid albumin gradient and evidence of bacterial peritonitis - high WBC count with high absolute neutrophil count in all five, isolation of Escherichia coli in three and Staphylococcus aureus in one. USG of patients with bacteremia also showed diffuse echo in intra-hepatic veins (Figures 4a & 4b).
Three patients were HBsAg positive with elevation of serum bilirubin and ALT. They were negative for acute viral hepatitis infection. Two of them had Escherichia coli bacteremia. All three patients were persistently positive for HBsAg since more than six months. All of them were positive for HBeAg, negative for anti-HBe and had very high HBVDNA levels. They were diagnosed with chronic hepatitis (CHB) in immune tolerant phase, with ALT elevation due to bacteremia related acute exacerbation of HVCS, which responded to antibiotic treatment. Two CHB patients with recurrent fever and jaundice developed liver cirrhosis. Development of liver cirrhosis in these patients was considered to be related to HVCS, as their clinical profile was consistent with the natural history of HVCS rather than with CHB.
A total of 49 (33 males and 16 females) children out of 178 developed liver cirrhosis (27.5%). Liver cirrhosis detected by USG was confirmed by liver biopsy in three children and direct inspection of the liver in another three. None of the patients with liver cirrhosis had evidence of Wilson’s disease. Among patients with liver cirrhosis, 63% had hepatomegaly, 44% had splenomegaly and 56% had dilated superficial veins with upward flow in abdomen and chest wall. Most of the patients with liver cirrhosis (83.6%) belonged to the 6 - 14 years age group. Cirrhosis also occurred in five children (10.2%) below one year of age (Table 3). Prevalence of liver cirrhosis was higher in males (31.4% vs. 21.4%). Among those who developed cirrhosis, six (12.2%) had complete obstruction of the IVC, 26 (53.0%) had moderate stenosis and remaining 17 (34.6%) had minimal stenosis. Recent and old organized thrombi were noted in all patients along the posterior wall of the IVC, close to the site of caval lesion.. The mean follow-up period of patients who developed liver cirrhosis was six months (range: 1- 9 months) in 21 and 4.9 years (range: 2-16 years) in the remaining 28 patients. Cirrhosis was detected at 3.8 months (range: 1-9 months) in 28 and 3.4 years (range: 1-10 years) in the remaining 21 patients. Liver cirrhosis followed acute severe exacerbation in 18 and recurrent acute exacerbations in 31.
Aerobic blood culture was done in 71 patients who initially presented with fever and had not received antibiotics. Bacteremia was detected in 43 (60.5%) patients. Gram negative organisms were detected in 40, mainly Escherichia coli and Klebsiella species. Staphylococcus aureus was detected in three cases. Ascitic fluid was examined in five patients. It showed high protein, high serum-ascitic fluid albumin gradient and evidence of bacterial peritonitis - high WBC count with high absolute neutrophil count in all five, isolation of Escherichia coli in three and Staphylococcus aureus in one. USG of patients with bacteremia also showed diffuse echo in intra-hepatic veins (Figures 4a & 4b).
Three patients were HBsAg positive with elevation of serum bilirubin and ALT. They were negative for acute viral hepatitis infection. Two of them had Escherichia coli bacteremia. All three patients were persistently positive for HBsAg since more than six months. All of them were positive for HBeAg, negative for anti-HBe and had very high HBVDNA levels. They were diagnosed with chronic hepatitis (CHB) in immune tolerant phase, with ALT elevation due to bacteremia related acute exacerbation of HVCS, which responded to antibiotic treatment. Two CHB patients with recurrent fever and jaundice developed liver cirrhosis. Development of liver cirrhosis in these patients was considered to be related to HVCS, as their clinical profile was consistent with the natural history of HVCS rather than with CHB.
A total of 49 (33 males and 16 females) children out of 178 developed liver cirrhosis (27.5%). Liver cirrhosis detected by USG was confirmed by liver biopsy in three children and direct inspection of the liver in another three. None of the patients with liver cirrhosis had evidence of Wilson’s disease. Among patients with liver cirrhosis, 63% had hepatomegaly, 44% had splenomegaly and 56% had dilated superficial veins with upward flow in abdomen and chest wall. Most of the patients with liver cirrhosis (83.6%) belonged to the 6 - 14 years age group. Cirrhosis also occurred in five children (10.2%) below one year of age (Table 3). Prevalence of liver cirrhosis was higher in males (31.4% vs. 21.4%). Among those who developed cirrhosis, six (12.2%) had complete obstruction of the IVC, 26 (53.0%) had moderate stenosis and remaining 17 (34.6%) had minimal stenosis. Recent and old organized thrombi were noted in all patients along the posterior wall of the IVC, close to the site of caval lesion.. The mean follow-up period of patients who developed liver cirrhosis was six months (range: 1- 9 months) in 21 and 4.9 years (range: 2-16 years) in the remaining 28 patients. Cirrhosis was detected at 3.8 months (range: 1-9 months) in 28 and 3.4 years (range: 1-10 years) in the remaining 21 patients. Liver cirrhosis followed acute severe exacerbation in 18 and recurrent acute exacerbations in 31.
 Patients continued to experience acute exacerbations after development of liver cirrhosis, though and responded to medical treatment. In between exacerbations they remained well. The frequency of acute exacerbations declined during followup even in those with cirrhosis, because parents paid more attention to nutrition, hygiene and early treatment of infections. Out of the 178 children two patients died, one from septicemia, ascites and renal failure within a month of diagnosis and the second child after 4 years follow-up, due to variceal bleeding and ascites related to acute exacerbations. Medical treatment of HVCS with high dose prolonged antibiotics was effective in children as compared to those treated surgically[21] or with anticoagulants.[37] Of the 18 children treated surgically by Gentil- Kocher et al in 1988, five died in the post-operative period and half of those who survived developed secondary thrombosis of the IVC or stenosis of the shunt during follow-up. Patients treated with anticoagulants had no decrease in mortality but had higher incidence of complications.
Discussion
HVCS was earlier considered a congenital vascular malformation.[14,15,38] The acquired nature of the disease and its relation to IVC thrombosis was first recognized by Okuda and colleagues.[39,40] It is now understood that bacterial infection causes thrombosis in the IVC.[41] Localized thrombosis/ thrombophlebitis of the IVC induced by bacterial infections at the site of hepatic vein entry is considered the initial lesion in HVCS, which on resolution leads to stenosis or complete obstruction.[42] Stenosis is frequently the early lesion observed in this disease,[12] and it can vary from a minimal localized thickening of the wall to severe narrowing. Fresh thrombosis on the initial lesion is frequently noted during acute exacerbations.[9,16,23] Acute exacerbations are precipitated by clinical or subclinical bacterial infections[23] which are often recurrent.[9,23] The IVC lesion eventually gets converted into a thick conical stenosis or complete obstruction. Thrombosis or endophlebitis of the intra-hepatic veins[21,23,26,27] also occurs during acute exacerbations. Vascular obstruction is followed by development of collaterals. Superficial cavo-caval collaterals are seen in the body trunk and deep collaterals are visualized by inferior veno cavogram (Figure 2). The deep collaterals have been extensively studied by injection technique in autopsies.[43] Intra-hepatic vein obstruction leads to development of collaterals within and around the liver, between the obstructed hepatic veins and the patent hepatic veins, portal vein or sub-capsular veins.[21,23]
HVCS is a cause of hepatic venous outflow obstruction (HVOO) similar to SOS caused by pyrrolizidine alkaloid or myeloablative therapy[44] and BCS caused by inherited or acquired thrombogenic conditions.[45] Both SOS and BCS are rare. There is however a wide overlap in the sites of BCS and HVCS vascular lesions. For example in some rarer causes of BCS[46-49] thrombosis occurs in the IVC which resolves into a thin membrane. Similarly hepatic vein ostia are frequently involved in HVCS, as are intra-hepatic veins during acute exacerbations, which resolve with stenosis or membrane formation. Given the similarities some authorities group HVCS with BCS.[50] The etiology, epidemiology, natural history and management of the two syndromes are however different. Briefly, whereas HVCS is a chronic disease related to bacterial infection that occurs in all age groups, predominantly in developing countries; BCS is related to thrombogenic conditions seen mainly in females of third decade, the outcome of symptomatic BCS being poor if not treated optimally with long-term anticoagulation, porto-systemic shunting or liver transplantation.[45]
HVCS in Nepal was seen in children from poor socioeconomic background and with history of recurrent bacterial infection. It affected children predominantly of 5 to 15 years age-group who presented with recurrent jaundice or ascites. It was complicated by liver cirrhosis in 27% patients. Cirrhosis developed within a few months to few years after onset of disease. The development of liver cirrhosis in HVCS was related to the severity and frequency of acute exacerbations and not directly either to the duration of the disease or to the severity of the caval lesion. Acute exacerbations were associated with fever, neutrophil leukocytosis, bacteremia in 60% of the patients, and USG evidence of fresh thrombus in the IVC along with echoic intra-hepatic veins. In adults also the clinical manifestations, development of liver cirrhosis and early death from septicemia, renal failure or variceal bleeding in HVCS, was related to acute exacerbations.[23] The progression and outcome of HVCS was thus determined by recurrent bacterial infections and not by the type or severity of the caval lesion. Pleasants made a similar observation in 1911 after reviewing a large number of cases, that once circulatory equilibrium was reestablished with development of collaterals, the duration of patient’s life was unaffected by the IVC occlusion per se. Authors who had observed long-term survival on conservative treatment in large series of patients from Japan[8] and Nepal[51] had raised doubts on the need of surgical or radiological correction of the caval lesions. Surgery in symptomatic cases was done to relieve hepatic venous congestion. But the chronic insidious course of the disease with occasional presentation with features of hepatic congestion like ascites, hepatomegaly or variceal bleeding suggests that the hepatic congestion is not precipitated by the caval obstruction but by the thrombosis occurring due to acute exacerbations. Surgery is not without complications and recurrence of the lesion after surgery is not uncommon.[21,29,52] Long-term histological follow-up after surgery showed no decrease in liver fibrosis or abrogation of cirrhosis in most of the patients.[21] Early medical treatment with prolonged high dose oral antibiotics, with diuretics when necessary, was found effective for the management of acute exacerbations in HVCS. This is evident by the low mortality among our patients.
Acute exacerbations are associated with fresh thrombosis in the IVC[9,16,23] and thrombosis or thrombophlebitis of the intrahepatic veins.[21,23,26] When thrombosis occurs close to the hepatic vein ostia, it causes HVOO resulting in hepatomegaly and ascites. Extensive ischemic necrosis of hepatocytes
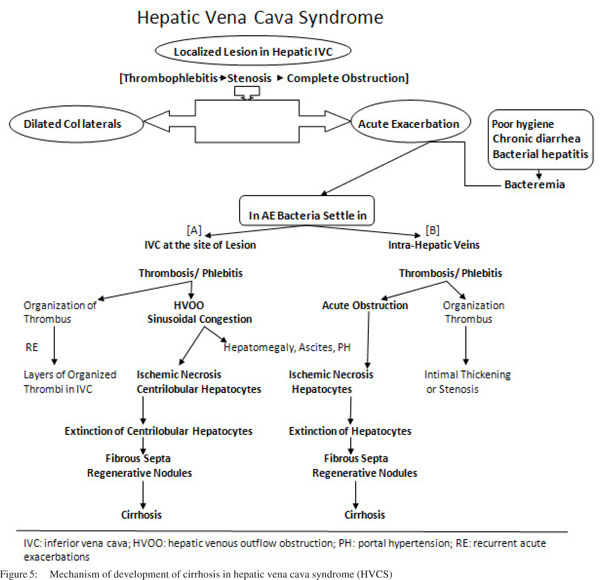
following thrombosis of sublobular hepatic vein[26] and consequent presence of recanalized thrombosed veins, intimal thickening or stenosis of intra-hepatic veins has been reported in autopsy studies.[21,53] Based on these observations two possible pathogenic mechanisms (Figure 5) of HVCS associated cirrhosis have been suggested:23 1) HVOO: obstruction of a large part of the hepatic outflow tract by the IVC thrombus (Figures 1b & 1c) results in sudden increase in the sinusoidal pressure followed by reflex reduction of hepatic arterial blood flow. The combined effect causes sinusoidal congestion and hemorrhage in the space of Disse; and ischemic necrosis of hepatocytes (Figure 6a) around the central vein. Apoptosis of the hepatocytes in the congested region is followed by fibrosis and formation of regenerative nodules, resulting in venocentric or reversed lobulation cirrhosis within a few months.[54] 2) Thrombosis or endophlebitis of the intra-hepatic veins (Figures 4a & 6b): obstruction of the intra-hepatic veins cause extinction of large areas of hepatocytes. This is followed by development of fibrous septa within a few weeks which subdivide the hepatic parenchyma into hepatocellular nodules and result in cirrhosis.[55] The obstruction of a hepatic vein branch is much more injurious. The obstruction of portal vein radicals alone, leads to atrophy but not extinction because ischemic insult is compensated by the arterial flow and there is minimal or no injury to the sinusoids.[55] Development of liver cirrhosis in steatohepatitis and chronic viral infection has also been considered to be due to obstruction of small hepatic veins adjacent to hepatic necroinflammation.[56] The mechanism of cirrhosis in HVCS is thus more explicit: ischemic necrosis and extinction of hepatic parenchyma secondary to obstruction of the hepatic vein and sinusoids by thrombosis or phlebitis during acute exacerbations.
Patients continued to experience acute exacerbations after development of liver cirrhosis, though and responded to medical treatment. In between exacerbations they remained well. The frequency of acute exacerbations declined during followup even in those with cirrhosis, because parents paid more attention to nutrition, hygiene and early treatment of infections. Out of the 178 children two patients died, one from septicemia, ascites and renal failure within a month of diagnosis and the second child after 4 years follow-up, due to variceal bleeding and ascites related to acute exacerbations. Medical treatment of HVCS with high dose prolonged antibiotics was effective in children as compared to those treated surgically[21] or with anticoagulants.[37] Of the 18 children treated surgically by Gentil- Kocher et al in 1988, five died in the post-operative period and half of those who survived developed secondary thrombosis of the IVC or stenosis of the shunt during follow-up. Patients treated with anticoagulants had no decrease in mortality but had higher incidence of complications.
Discussion
HVCS was earlier considered a congenital vascular malformation.[14,15,38] The acquired nature of the disease and its relation to IVC thrombosis was first recognized by Okuda and colleagues.[39,40] It is now understood that bacterial infection causes thrombosis in the IVC.[41] Localized thrombosis/ thrombophlebitis of the IVC induced by bacterial infections at the site of hepatic vein entry is considered the initial lesion in HVCS, which on resolution leads to stenosis or complete obstruction.[42] Stenosis is frequently the early lesion observed in this disease,[12] and it can vary from a minimal localized thickening of the wall to severe narrowing. Fresh thrombosis on the initial lesion is frequently noted during acute exacerbations.[9,16,23] Acute exacerbations are precipitated by clinical or subclinical bacterial infections[23] which are often recurrent.[9,23] The IVC lesion eventually gets converted into a thick conical stenosis or complete obstruction. Thrombosis or endophlebitis of the intra-hepatic veins[21,23,26,27] also occurs during acute exacerbations. Vascular obstruction is followed by development of collaterals. Superficial cavo-caval collaterals are seen in the body trunk and deep collaterals are visualized by inferior veno cavogram (Figure 2). The deep collaterals have been extensively studied by injection technique in autopsies.[43] Intra-hepatic vein obstruction leads to development of collaterals within and around the liver, between the obstructed hepatic veins and the patent hepatic veins, portal vein or sub-capsular veins.[21,23]
HVCS is a cause of hepatic venous outflow obstruction (HVOO) similar to SOS caused by pyrrolizidine alkaloid or myeloablative therapy[44] and BCS caused by inherited or acquired thrombogenic conditions.[45] Both SOS and BCS are rare. There is however a wide overlap in the sites of BCS and HVCS vascular lesions. For example in some rarer causes of BCS[46-49] thrombosis occurs in the IVC which resolves into a thin membrane. Similarly hepatic vein ostia are frequently involved in HVCS, as are intra-hepatic veins during acute exacerbations, which resolve with stenosis or membrane formation. Given the similarities some authorities group HVCS with BCS.[50] The etiology, epidemiology, natural history and management of the two syndromes are however different. Briefly, whereas HVCS is a chronic disease related to bacterial infection that occurs in all age groups, predominantly in developing countries; BCS is related to thrombogenic conditions seen mainly in females of third decade, the outcome of symptomatic BCS being poor if not treated optimally with long-term anticoagulation, porto-systemic shunting or liver transplantation.[45]
HVCS in Nepal was seen in children from poor socioeconomic background and with history of recurrent bacterial infection. It affected children predominantly of 5 to 15 years age-group who presented with recurrent jaundice or ascites. It was complicated by liver cirrhosis in 27% patients. Cirrhosis developed within a few months to few years after onset of disease. The development of liver cirrhosis in HVCS was related to the severity and frequency of acute exacerbations and not directly either to the duration of the disease or to the severity of the caval lesion. Acute exacerbations were associated with fever, neutrophil leukocytosis, bacteremia in 60% of the patients, and USG evidence of fresh thrombus in the IVC along with echoic intra-hepatic veins. In adults also the clinical manifestations, development of liver cirrhosis and early death from septicemia, renal failure or variceal bleeding in HVCS, was related to acute exacerbations.[23] The progression and outcome of HVCS was thus determined by recurrent bacterial infections and not by the type or severity of the caval lesion. Pleasants made a similar observation in 1911 after reviewing a large number of cases, that once circulatory equilibrium was reestablished with development of collaterals, the duration of patient’s life was unaffected by the IVC occlusion per se. Authors who had observed long-term survival on conservative treatment in large series of patients from Japan[8] and Nepal[51] had raised doubts on the need of surgical or radiological correction of the caval lesions. Surgery in symptomatic cases was done to relieve hepatic venous congestion. But the chronic insidious course of the disease with occasional presentation with features of hepatic congestion like ascites, hepatomegaly or variceal bleeding suggests that the hepatic congestion is not precipitated by the caval obstruction but by the thrombosis occurring due to acute exacerbations. Surgery is not without complications and recurrence of the lesion after surgery is not uncommon.[21,29,52] Long-term histological follow-up after surgery showed no decrease in liver fibrosis or abrogation of cirrhosis in most of the patients.[21] Early medical treatment with prolonged high dose oral antibiotics, with diuretics when necessary, was found effective for the management of acute exacerbations in HVCS. This is evident by the low mortality among our patients.
Acute exacerbations are associated with fresh thrombosis in the IVC[9,16,23] and thrombosis or thrombophlebitis of the intrahepatic veins.[21,23,26] When thrombosis occurs close to the hepatic vein ostia, it causes HVOO resulting in hepatomegaly and ascites. Extensive ischemic necrosis of hepatocytes
following thrombosis of sublobular hepatic vein[26] and consequent presence of recanalized thrombosed veins, intimal thickening or stenosis of intra-hepatic veins has been reported in autopsy studies.[21,53] Based on these observations two possible pathogenic mechanisms (Figure 5) of HVCS associated cirrhosis have been suggested:23 1) HVOO: obstruction of a large part of the hepatic outflow tract by the IVC thrombus (Figures 1b & 1c) results in sudden increase in the sinusoidal pressure followed by reflex reduction of hepatic arterial blood flow. The combined effect causes sinusoidal congestion and hemorrhage in the space of Disse; and ischemic necrosis of hepatocytes (Figure 6a) around the central vein. Apoptosis of the hepatocytes in the congested region is followed by fibrosis and formation of regenerative nodules, resulting in venocentric or reversed lobulation cirrhosis within a few months.[54] 2) Thrombosis or endophlebitis of the intra-hepatic veins (Figures 4a & 6b): obstruction of the intra-hepatic veins cause extinction of large areas of hepatocytes. This is followed by development of fibrous septa within a few weeks which subdivide the hepatic parenchyma into hepatocellular nodules and result in cirrhosis.[55] The obstruction of a hepatic vein branch is much more injurious. The obstruction of portal vein radicals alone, leads to atrophy but not extinction because ischemic insult is compensated by the arterial flow and there is minimal or no injury to the sinusoids.[55] Development of liver cirrhosis in steatohepatitis and chronic viral infection has also been considered to be due to obstruction of small hepatic veins adjacent to hepatic necroinflammation.[56] The mechanism of cirrhosis in HVCS is thus more explicit: ischemic necrosis and extinction of hepatic parenchyma secondary to obstruction of the hepatic vein and sinusoids by thrombosis or phlebitis during acute exacerbations.
Liver cirrhosis in HVCS is characterized by some distinctive features: i) cirrhosis occur at early stage of the disease, ii) it has better prognosis if infection is prevented and adequately treated because liver damage in this disease is episodic and related to the acute exacerbations, iii) unlike other causes of liver cirrhosis where the hepatic veins appear attenuated on USG, in HVCS they are frequently dilated, iv) liver USG in HVCS related liver cirrhosis may show echoic intra-hepatic veins, calcified foci and large regenerative nodules. The gall bladder may be thick and edematous during acute exacerbations, v) liver cirrhosis due to other causes witnesses complications like ascites, jaundice, edema of legs[57] and bacterial infection[58] late in the course of illness with the development of hepatic decompensation. These also constitute the indications for liver transplantation as they predict poor prognosis. In HVCS these symptoms occur before the development of cirrhosis or during early liver cirrhosis. It is related to HVOO, which is caused by bacterial infection-induced thrombosis and responds to medical treatment.
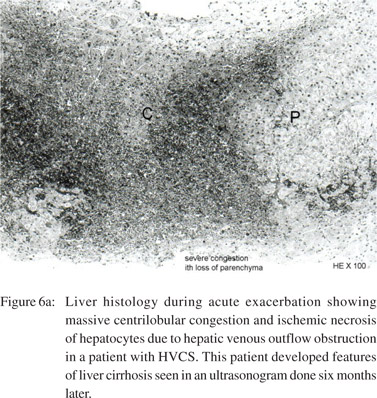
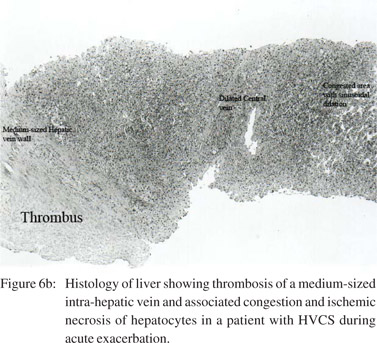 The onset of liver cirrhosis due to other liver diseases is clinically marked by the development of ascites, esophageal varices or splenomegaly. In HVCS these clinical features occur early even before development of liver cirrhosis. Serial followup USG has proven helpful in early detection of HVCS associated liver cirrhosis.[23] This combined with serial followup blood counts for thrombocytopenia or leucopenia increase the sensitivity of diagnosis. In our study the ultrasonographic diagnosis of liver cirrhosis was confirmed by liver biopsy in three patients and by direct observation of the liver surface in another three. Liver biopsy in two patients after episodes of fever, jaundice and ascites but without USG features of liver cirrhosis (Figures 6a & 6b) showed changes in liver compatible with HVCS without liver cirrhosis. Our previous study showed that although serial USG diagnoses liver cirrhosis later in the course of disease when compared to serial liver biopsy, but the diagnosis is specific.[23] Although USG is operator dependent, it has the advantages of being a non-invasive test, with low cost, good reproducibility and easy availability in developing countries where HVCS is endemic. Furthermore, USG evidence can be supported with a liver biopsy where necessary. Studies suggests that USG is reliable for the diagnosis of liver cirrhosis,[59] especially in children.[60] In our experience USG is practical, cost-effective and provides detailed information about the liver and IVC in HVCS patients.
In summary, HVCS is characterized by recurrent thrombosis/phlebitis of the IVC and intra-hepatic veins during acute exacerbations. Given its characteristic location in the IVC just opposite the hepatic vein opening, thrombosis at this site frequently results in HVOO. HVOO and thrombosis/phlebitis of the medium-sized intra-hepatic veins are followed by ischemic necrosis of hepatocytes and development of liver cirrhosis. Serial USG preferably done by the treating physician can help monitor the changes in liver and the IVC during acute exacerbations and aid in early detection of liver cirrhosis. Despite the rapid economic growth in Asia, a large segment of population in Asia and Africa still live in conditions of
suboptimal hygiene, where HVCS may be a common yet preventable cause of liver disease and cirrhosis. Early diagnosis of HVCS requires early recognition of the IVC lesion[61] by USG before development of obstruction. Awareness of the condition and the use of USG to examine the IVC and liver in patients with bacterial infection and/or ALT elevation are likely to widen the diagnostic net and provide better opportunity to prevent cirrhosis in these patients.
References
The onset of liver cirrhosis due to other liver diseases is clinically marked by the development of ascites, esophageal varices or splenomegaly. In HVCS these clinical features occur early even before development of liver cirrhosis. Serial followup USG has proven helpful in early detection of HVCS associated liver cirrhosis.[23] This combined with serial followup blood counts for thrombocytopenia or leucopenia increase the sensitivity of diagnosis. In our study the ultrasonographic diagnosis of liver cirrhosis was confirmed by liver biopsy in three patients and by direct observation of the liver surface in another three. Liver biopsy in two patients after episodes of fever, jaundice and ascites but without USG features of liver cirrhosis (Figures 6a & 6b) showed changes in liver compatible with HVCS without liver cirrhosis. Our previous study showed that although serial USG diagnoses liver cirrhosis later in the course of disease when compared to serial liver biopsy, but the diagnosis is specific.[23] Although USG is operator dependent, it has the advantages of being a non-invasive test, with low cost, good reproducibility and easy availability in developing countries where HVCS is endemic. Furthermore, USG evidence can be supported with a liver biopsy where necessary. Studies suggests that USG is reliable for the diagnosis of liver cirrhosis,[59] especially in children.[60] In our experience USG is practical, cost-effective and provides detailed information about the liver and IVC in HVCS patients.
In summary, HVCS is characterized by recurrent thrombosis/phlebitis of the IVC and intra-hepatic veins during acute exacerbations. Given its characteristic location in the IVC just opposite the hepatic vein opening, thrombosis at this site frequently results in HVOO. HVOO and thrombosis/phlebitis of the medium-sized intra-hepatic veins are followed by ischemic necrosis of hepatocytes and development of liver cirrhosis. Serial USG preferably done by the treating physician can help monitor the changes in liver and the IVC during acute exacerbations and aid in early detection of liver cirrhosis. Despite the rapid economic growth in Asia, a large segment of population in Asia and Africa still live in conditions of
suboptimal hygiene, where HVCS may be a common yet preventable cause of liver disease and cirrhosis. Early diagnosis of HVCS requires early recognition of the IVC lesion[61] by USG before development of obstruction. Awareness of the condition and the use of USG to examine the IVC and liver in patients with bacterial infection and/or ALT elevation are likely to widen the diagnostic net and provide better opportunity to prevent cirrhosis in these patients.
References
- Kiyosawa K, Sodeyama T, Tanaka E, Gibo Y, Yoshizawa K, Nakano Y, et al. Interrelationship of blood transfusion, non-A, non-B hepatitis and hepatocellular carcinoma: analysis by detection of antibody to hepatitis C virus. Hepatology. 1990;12:671–5.
- Mieli-Vergani G, Vergani D. Approach to a child with chronic liver disease. J Gastroenterol Hepatol. 2004;19:S377–8.
- Vajro P, Hadchouel P, Hadchouel M, Bernard O, Alagille D. Incidence of cirrhosis in children with chronic hepatitis. J Pediatr. 1990;117:392–6.
- Bras G, Brooks SE, Watler DC. Cirrhosis of the liver in Jamaica. J Pathol Bacteriol. 1961;82:503–12.
- Nayak NC. Indian childhood cirrhosis. In: Ahuja MMS, editor. Progress in clinical medicine. Second series, Arnold –Heinnemann; 1978. p.304–14.
- Pandit A. Indian childhood cirrhosis: a reappraisal. In: Advances in liver diseases. Tandon BN, Nayak NC, Nundy S, editors. New Delhi: Macmillan India; 1989.p.69–74.
- Shrestha SM, Okuda K, Uchida T, Maharjan KG, Shrestha S, Joshi BL, et al. Endemicity and clinical picture of liver disease due to obstruction of the hepatic portion of the inferior vena cava in Nepal. J Gastroenterol Hepatol. 1996;11:170–9.
- Nakamura T, Nakamura S, Aikawa T, Suzuki O, Onodera A, Karoji N. Obstruction of the inferior vena cava in the hepatic portion and the hepatic veins. Report of eight cases and review of the Japanese literature. Angiology. 1968;19:479–98.
- Takeuchi J, Takada A, Hasumura Y, Matsuda Y, Ikegami P. Budd- Chiari syndrome associated with obstruction of the inferior vena cava. A report of seven cases. Am J Med. 1971;51:11–20.
- Nakamura S, Takezawa Y. Obstruction of the inferior vena cava in the hepatic portion and hepatocellular carcinoma. Tohoku J Exp Med. 1982;138:119–20.
- Datta DV, Saha S, Singh SA, Gupta BB, Aikat BK, Chhuttani PN. Clinical spectrum of Budd-Chiari syndrome in Chandigrah with particular reference to obstruction of intrahepatic portion of inferior vena cava. Indian J Med Res. 1972;60:385–402.
- Madangopalan N, Solomon V, Jayanthi V, Raghuram K, Balakumar M, Kandasway I, et al. Clinical spectrum of chronic Budd-Chiari syndrome and surgical relief for ‘coarctation’ of the inferior vena cava. J Gastroenterol Hepatol. 1986;1:359–69.
- Dilawari JB, Bambery P, Chawla Y, Kaur U, Bhusnurmath SR, Malhotra HS, et al. Hepatic outflow obstruction (Budd-Chiari syndrome). Experience with 177 patients and a review of the literature. Medicine (Baltimore). 1994;73:21–36.
- Simson IW. Membranous obstruction of the inferior vena cava and hepatocellular carcinoma in South Africa. Gastroenterology. 1982;82:171–8.
- Kew MC, McKnight A, Hodkinson J, Bukofzer S, Esser JD. The role of membranous obstruction of the inferior vena cava in the etiology of hepatocellular carcinoma in Southern African blacks. Hepatology. 1989;9:121–5.
- Thompson T, Turnbull HM. Primary occlusion of the ostia of the hepatic veins. Q J Med. 1912;5:277–95.
- Hutchison R, Simpson SL. Occlusion of the hepatic veins with cirrhosis of the liver. Arch Dis Child. 1930;5:167–86.
- Parker RG. Occlusion of the hepatic veins in man. Medicine (Baltimore). 1959;38:369–402.
- Shrestha S, Shrestha S. Bacterial peritonitis in hepatic inferior vena cava disease: a hypothesis to explain the cause of infection in high protein ascites. Hepatol Res. 2002;24:42.
- Shrestha SM. Pleural effusion in hepatic vena cava disease. Kathmandu Univ Med J (KUMJ). 2007;5:218–24.
- Gentil-Kocher S, Bernard O, Brunelle F, Hadchouel M, Maillard JN, Valayer J, et al. Budd-Chiari syndrome in children: report of 22 cases. J Pediatr. 1988;113:30–8.
- Arora A, Sharma MP, Acharya SK, Panda SK, Berry M. Diagnostic utility of ultrasonography in hepatic venous outflow tract obstruction in a tropical country. J Gastroenterol Hepatol. 1991;6:368–73.
- Shrestha SM. Liver cirrhosis and hepatocellular carcinoma in hepatic vena cava disease, a liver disease caused by obstruction of inferior vena cava. Hepatol Int. 2009;3:392–402.
- Wilks S. Obstruction of hepatic veins and vena cava by fibrous deposit in the liver. Trans Pathol Soc Lond. 1862;13:122–4.
- Osler W. Obliteration of Vena Cava Inferior, with great Stenosis of Orifices of Hepatic Veins. J Anat Physiol. 1879;13:291–304.
- Mann JD, Hall IW. Obstruction of inferior vena cava. Edinburgh Med J. 1904;16:56–62.
- Rigdon RH. On the relation between thrombophlebitis of inferior vena cava and occlusion of hepatic veins (Endophlebitis hepatic obliterans). Bull Johns Hopkins Hosp. 1933;53:162–71.
- Kibel MA, Marsden HB. Inferior vena caval and hepatic vein thrombosis: the Chiari syndrome in childhood. Arch Dis Child. 1956;31:225–8.
- Wang ZG, Zhang FJ, Li XQ, Meng QY. Management of Budd-Chiari syndrome: what is the best approach? J Gastroenterol Hepatol. 2004;19:S212–18.
- Kao WY, Hung HH, Lu HC, Lin HC, Wu JC, Lee SD, et al. Hepatocellular carcinoma with presentation of budd-Chiari syndrome. J Chin Med Assoc. 2010;73:93–6.
- Seo JB, Chung JW, Park JH, Kim SH, Kim TK, Han JK, et al. Benign obstruction of the hepatic inferior vena cava complicated by hepatocellular carcinoma: combined interventional management. AJR Am J Roentgenol. 1998;170:655–9.
- Awwad S. The Budd-Chiari syndrome. J Egypt Med Assoc. 1952;35:650–69.
- Okuda K. Membranous obstruction of the inferior vena cava: etiology and relation to hepatocellular carcinoma. Gastroenterology. 1982;82:376–9.
- Angelman H, Spencer R. The syndrome of obstruction of inferior vena cava in childhood. Br Med J. 1950;2:752–5.
- Taneja A, Mitra SK, Moghe PD, Rao PN, Samanta N, Kumar L. Budd-Chiari syndrome in childhood secondary to inferior vena caval obstruction. Pediatrics. 1979;63:808–12.
- Hoefs JC. Diagnostic paracentesis. A potent clinical tool. Gastroenterology. 1990;98:230–6.
- Shrestha SM. Non-surgical treatment of hepatic vena cava disease. In: Postgraduate course and current reviews. Sarin SK, Sharma BC, Kumar M, editors. New Delhi: CBS Publisher and Distributors; 2004.p.43–9.
- Hirooka M. Embryonic abnormality as the pathogenesis of membranous obliteration of the inferior vena cava in the hepatic portion. Acta Hepatol Jpn. 1969;10:566–77.
- Asahi Central Hospital. Aetiology of membranous obstruction of inferior vena cava: congenital or acquired? Gastroeneterol Int. 1990;3:70–80.
- Okuda K, Kage M, Shrestha SM. Proposal of a new nomenclature for Budd-Chiari syndrome: hepatic vein thrombosis versus thrombosis of the inferior vena cava at its hepatic portion. Hepatology. 1998;28:1191–8.
- Ceyhan M, Kanra G, Benderlioglu B, Secmeer G, Hicsonmez G, Kirazli S. Transient protein S deficiency with deep venous thrombosis during Salmonella typhimurium infection. Arch Dis Child. 1993;68:138–9.
- Shrestha SM, Shrestha S. Hepatic vena cava disease: Etiologic relation to bacterial infection. Hepatol Res. 2007;37:196–204.
- Pleasants JH. Obstruction of the inferior vena cava with report of eighteen cases. Johns Hopkins Hospital Report. 1911;16:363–548.
- DeLeve LD, Shulman HM, McDonald GB. Toxic injury to hepatic sinusoids: sinusoidal obstruction syndrome (veno-occlusive disease). Semin Liver Dis. 2002;22:27–42.
- Plessier A, Valla DC. Budd-Chiari syndrome. Semin Liver Dis. 2008;28:259–69.
- Blanshard C, Dodge G, Pasi J, Ormiston M, Dick R, Burroughs AK. Membranous obstruction of the inferior vena cava in a patient with factor V Leiden: evidence for a post-thrombotic aetiology. J Hepatol. 1997;26:731–5.
- Sevenet F, Deramond H, Hadengue A, Casadevall N, Delamarre J, Capron JP. Membranous obstruction of the inferior vena cava associated with a myeloproliferative disorder: a clue to membrane formation? Gastroenterology. 1989;97:1019–21.
- Terabayashi H, Okuda K, Nomura F, Ohnishi K, Wong P. Transformation of inferior vena caval thrombosis to membranous obstruction in a patient with the lupus anticoagulant. Gastroenterology. 1986;91:219–24.
- Bayraktar Y, Balkanci F, Bayraktar M, Calguneri M. Budd-Chiari syndrome: a common complication of Behcet’s disease. Am J Gastroenterol. 1997;92:858–62.
- Valla DC. Budd-Chiari syndrome. In: Zakim and Boyer’s hepatology: Text book of liver disease. Boyer TD, Wright TL, Manns MP, editors. Philadelphia: Saunders; 2006.p.877–96.
- Shrestha SM. Hepatic venous outflow obstruction in Nepal. Trop Gastroenterol. 1996;17:165–71.
- Kohli V, Pande GK, Dev V, Reddy KS, Kaul U, Nundy S. Management of hepatic venous outflow obstruction. Lancet. 1993;342:718–22.
- Kage M, Arakawa M, Kojiro M, Okuda K. Histopathology of membranous obstruction of the inferior vena cava in the Budd- Chiari syndrome. Gastroenterology. 1992;102:2081–90.
- Tanaka M, Wanless IR. Pathology of the liver in Budd-Chiari syndrome: portal vein thrombosis and the histogenesis of venocentric cirrhosis, veno-portal cirrhosis, and large regenerative nodules. Hepatology. 1998;27:488–96.
- Wanless IR. Pathogenesis of cirrhosis. J Gastroenterol Hepatol. 2004;19:S369–71.
- Wanless IR, Shiota K. The pathogenesis of nonalcoholic steatohepatitis and other fatty liver diseases: a four-step model including the role of lipid release and hepatic venular obstruction in the progression to cirrhosis. Semin Liver Dis. 2004;24:99–106.
- Wright TL, Boyer TD. Diagnosis and management of cirrhotic ascites. In: Hepatology. Zakim D, Boyer TD, editors. WB Saunders; 1990.p.616–34.
- Arvaniti V, D’Amico G, Fede G, Manousou P, Tsochatzis E, Pleguezuelo M, et al. Infections in patients with cirrhosis increase mortality four-fold and should be used in determining prognosis. Gastroenterology. 2010;139:1246–56.
- Kudo M, Zheng RQ, Kim SR, Okabe Y, Osaki Y, Iijima H, et al. Diagnostic accuracy of imaging for liver cirrhosis compared to histologically proven liver cirrhosis. A multicenter collaborative study. Intervirology. 2008;51 Suppl 1:17–26.
- Zhu JA, Hu B. Ultrasonography in predicting and screening liver cirrhosis in children: a preliminary study. World J Gastroenterol. 2003;9:2348–9.
- Shrestha SM, Joshi BL, Shrestha S, Maharajan KG. Cavographic study of an early stage of obstruction of the hepatic portion of the inferior vena cava. J Gastroenterol Hepatol. 2000;15:202–10.
|
|
|
|
|
|