|
|
|
|
 |
|
| |
 |
|
|
Quarterly Reviews |
|
|
|
|
|
Keywords :
idiopathic recurrent acute pancreatitis, microlithiasis, chronic pancreatitis, pancreas divisum |
|
|
Saurabh Kedia, Rajan Dhingra, Pramod Kumar Garg
Department of Gastroenterology,
All India Institute of Medical Sciences,
New Delhi, India
Corresponding Author:
Dr Pramod Kumar Garg
Email: pkgarg@aiims.ac.in
DOI:
http://dx.doi.org/10.7869/tg.116
Abstract
Recurrent acute pancreatitis (RAP) is defined as more than two attacks of acute pancreatitis (AP) without any evidence of underlying chronic pancreatitis (CP). As the known causes of AP are generally taken care of, RAP usually occurs in the idiopathic group, which forms 20%– 25% of cases of AP. The causes of idiopathic RAP (IRAP) can be mechanical, toxic–metabolic, anatomical, or miscellaneous. Microlithiasis commonly reported from the West is not a common cause of IRAP among Indian patients. Pancreas divisum (PD) is now believed as a cofactor, the main factor being associated genetic mutations. The role of Sphincter of Oddi dysfunction (SOD) as a cause of IRAP remains controversial. Malignancy should be ruled out in any patient with IRAP >50 years of age. Early CP can present initially as RAP. The work-up of patients with IRAP includes a detailed history and investigations. Primary investigations include liver function tests (LFT), serum calcium and triglyceride, abdominal ultrasonography (USG) and contrast-ehhanced computed tomography (CECT) abdomen. Endoscopic ultrasound (EUS), magnetic resonance cholangiopancreatography (MRCP) and possibly endoscopic retrograde cholangiopancreatography (ERCP) are indicated in the secondary phase if the work-up is negative after the primary investigations. EUS is advised usually 6–8 weeks after an acute episode. Treatment of patients with IRAP is aimed at the specific aetiology. In general, empirical cholecystectomy should be discouraged with the availability and widespread use of EUS. Endoscopic sphincterotomy is advised if there is strong suspicion of SOD. Minor papilla sphincterotomy should be carried out in those with PD but with limited expectations. Regular follow-up of patients with IRAP is necessary because most patients are likely to develop CP in due course.
|
48uep6bbphidvals|601 48uep6bbph|2000F98CTab_Articles|Fulltext Acute pancreatitis (AP) is a common clinical problem in gastrointestinal practice. It is diagnosed in the presence of acute onset of upper abdominal pain, elevated amylase and/or lipase levels, and imaging evidence of pancreatic and peripancreatic inflammation. Its incidence is increasing over the past 2 decades and varies from 30 to 80 per 100,000population.[1–4] In the USA, AP ranked first among all the gastrointestinal and hepatology-related hospitalizations in the year 2009 and accounted for 274,119 admissions annually with treatment costs exceeding $2.5 billion.[5]
AP is either interstitial or necrotizing. Acute interstitial pancreatitis is seen in 70%–80% of patients and runs a mild course. In contrast, acute necrotizing pancreatitis—a severe form of the disease—is present in 20%–30% of patients and associated with a mortality rate of up to 40%.[6] Others and we have shown that the extent of pancreatic necrosis correlates with organ failure and mortality.[7,8]
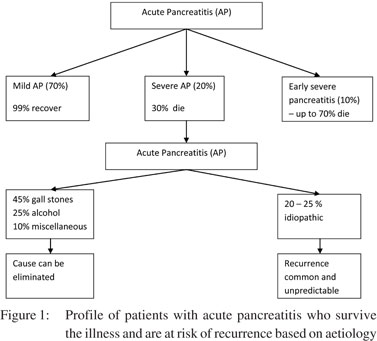
The cause of AP is evident after standard investigations in about 70%–80% of patients during or after the first attack. Gallstones are the cause of AP in about 45%, alcohol intake in 20%–25%, post-endoscopic retrograde cholangiopancreatography (ERCP) in 5%–7%, and miscellaneous in about 5% of cases.[9] Thus, the cause is not evident in 20%–25% of patients after standard initial evaluation and such patients are labelled as having idiopathic AP (Figure 1). The standard initial evaluation includes a detailed history and physical examination, and investigations such as liver function tests (LFTs), lipid profile, serum calcium and imaging.
 Risk of recurrence after an attack of AP
Most patients who have had an attack of AP are at risk of having a recurrence of pancreatitis if the offending cause/agent is not removed/eliminated. Of the common causes of AP, gallstones and alcohol are most likely to cause recurrent pancreatitis. Thus, the risk of recurrence is often predictable and there is a window of opportunity for treating the cause to reduce recurrence. Fortunately, most causes of AP can be removed/eliminated/treated, e.g. gallstones, alcohol intake, drugs, trauma, ERCP, etc. However, the risk of recurrence in patients with idiopathic pancreatitis is difficult to predict.
Recurrent acute pancreatitis (RAP)
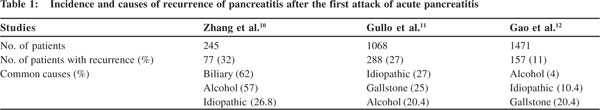
Recurrent acute pancreatitis (RAP) is defined as more than two attacks of AP without any evidence of underlying chronic pancreatitis (CP). The studies which have looked at the incidence of recurrence in respect of the aetiology of AP are summarized in Table 1. However, the true incidence and aetiology of RAP in the idiopathic group are not clear from these studies as the commonest causes of RAP in these series were alcohol intake and gallstones.[10–12] Patients with gallstones undergo cholecystectomy and thus there is no further risk of recurrence in the majority of patients while patients who continue to consume alcohol usually develop CP. In general, as the known causes of AP are taken care of, RAP usually occurs in the idiopathic group. This article will focus on idiopathic RAP (IRAP) which is the most difficult group of patients because of problems in identifying its possible cause, uncertainty about its natural course in terms of recurrence, and long-term outcome.
Aetiology of idiopathic RAP (IRAP)
The causes of IRAP can be grouped into mechanical, toxic– metabolic, anatomical, and miscellaneous Table 2.
Risk of recurrence after an attack of AP
Most patients who have had an attack of AP are at risk of having a recurrence of pancreatitis if the offending cause/agent is not removed/eliminated. Of the common causes of AP, gallstones and alcohol are most likely to cause recurrent pancreatitis. Thus, the risk of recurrence is often predictable and there is a window of opportunity for treating the cause to reduce recurrence. Fortunately, most causes of AP can be removed/eliminated/treated, e.g. gallstones, alcohol intake, drugs, trauma, ERCP, etc. However, the risk of recurrence in patients with idiopathic pancreatitis is difficult to predict.
Recurrent acute pancreatitis (RAP)
Recurrent acute pancreatitis (RAP) is defined as more than two attacks of AP without any evidence of underlying chronic pancreatitis (CP). The studies which have looked at the incidence of recurrence in respect of the aetiology of AP are summarized in Table 1. However, the true incidence and aetiology of RAP in the idiopathic group are not clear from these studies as the commonest causes of RAP in these series were alcohol intake and gallstones.[10–12] Patients with gallstones undergo cholecystectomy and thus there is no further risk of recurrence in the majority of patients while patients who continue to consume alcohol usually develop CP. In general, as the known causes of AP are taken care of, RAP usually occurs in the idiopathic group. This article will focus on idiopathic RAP (IRAP) which is the most difficult group of patients because of problems in identifying its possible cause, uncertainty about its natural course in terms of recurrence, and long-term outcome.
Aetiology of idiopathic RAP (IRAP)
The causes of IRAP can be grouped into mechanical, toxic– metabolic, anatomical, and miscellaneous Table 2.
 Mechanical causes
Mechanical causes
Microlithiasis
Microliths are defined as gallstones <3 mm in size. A cut-off of 3 mm has been taken because abdominal ultrasound (USG) can diagnose gallstones >3 mm in size. The only difference between gallstones and microliths is in their size and hence the pathophysiology of microliths is same as that of gallstones.[13]
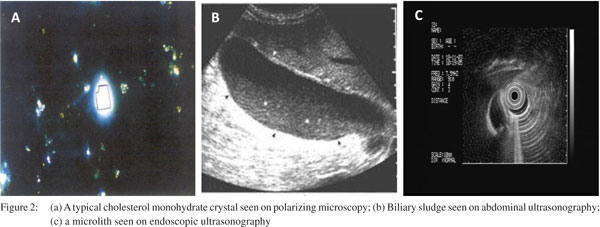
Therefore, microliths are expected to increase in size and number in their natural course. Indeed, studies on model bile have shown that there is an increase of 15% per week in the size of gallstones which plateau in a week, and resume their grow when bile is replaced by fresh bile. Therefore, microliths would finally develop into macroliths, i.e. detectable gallstones in the presence of lithogenic bile. Ros et al.[14] had shown that about 92% of patients with idiopathic pancreatitis and suspected microlithiasis (presence of cholesterol crystals in the bile) were found to have detectable gallstones on follow-up abdominal USG suggesting their natural increase in size. Similarly, in another study 75% of patients developed gallstones on followup.[15] Microliths are predominantly composed of cholesterol. The diagnosis of microlithiasis can be suspected on bile microscopy which might show cholesterol monohydrate, calcium bilirubinate, and/or calcium carbonate crystals (Figure 2a). Microlithiasis should not be confused with biliary sludge which is a diagnosis made on abdominal USG as lowamplitude echoes in the gallbladder without shadowing and that layer in the dependent part of the gallbladder. Biliary sludge is composed of a suspension of crystals, mucin, glycoproteins, cellular debris and proteinaceous material (Figure 2b). However, both the microlithiasis and biliary sludge are used interchangeably in the context of idiopathic pancreatitis.
Diagnosis of microlithiasis
Bile microscopy has a sensitivity of 65%–95% for detecting biliary crystals which could be an indirect evidence for microliths.[16] For bile microscopy, duodenal bile is obtained after cholecystokinin analogue or rapid amino acid infusion, centrifuged and the sediment is examined under polarizing microscope as shown by us previously.[17] Presence of more than three crystals per slide is taken as suggestive of microliths. However, an important caveat is that bile microscopy detects biliary crystals and not microliths and is an indirect indicator of the presence of microliths. Crystals and microliths are not equivalent, and it is the microlith which can cause pancreatitis but not the crystal which is microscopic in size. Bile microscopy can also be performed on bile from the common bile duct (CBD) obtained during ERCP. In that case, bile should be aspirated before contrast injection. Buscail et al.[17] showed that in 41/50 patients with gallstones and 7/22 patients with suspected microliths, CBD bile was positive for biliary crystals. However, microliths are best diagnosed on endoscopic ultrasound (EUS), which has a sensitivity of 96% for diagnosing microliths (Figure 2c).[18]
 Microlithiasis and pancreatitis: Microliths by virtue of their small size are likely to pass through the cystic duct and may lead to pancreatitis by causing transient obstruction at the ampulla of Vater due to impaction of the stone.[19] Just as in gallstone pancreatitis, microliths as a cause of AP should be suspected as the cause of pancreatitis in the presence of abnormal LFT within 24–48 hours of the onset of AP. In a study, abnormal LFT has about 84% sensitivity and specificity to diagnose biliary pancreatitis.[20] The prevalence of microlithiasis as the cause of RAP has varied across various studies (Table 3). Few studies have shown a high prevalence of microlithiasis (50%–73%) as the cause of IRAP.[14,15,21,22] On the other hand, many studies including a study from our centre have shown a low prevalence of microlithiasis ranging from 6%–16%.[23–25] In a study of 75 patients with IRAP, we found that only 10 (13%) patients had microlithiasis, 35 (47%) patients developed CP during the follow-up—10 (13%) patients had miscellaneous aetiologies and no cause was found in the remaining 18 (24%) patients. Ten of these 35 patients who developed CP had biliary microlithiasis—8 of these 10 patients had undergone cholecystectomy/endoscopic sphincterotomy, yet continued to have recurrent pancreatitis and developed CP.[25]
Microlithiasis and pancreatitis: Microliths by virtue of their small size are likely to pass through the cystic duct and may lead to pancreatitis by causing transient obstruction at the ampulla of Vater due to impaction of the stone.[19] Just as in gallstone pancreatitis, microliths as a cause of AP should be suspected as the cause of pancreatitis in the presence of abnormal LFT within 24–48 hours of the onset of AP. In a study, abnormal LFT has about 84% sensitivity and specificity to diagnose biliary pancreatitis.[20] The prevalence of microlithiasis as the cause of RAP has varied across various studies (Table 3). Few studies have shown a high prevalence of microlithiasis (50%–73%) as the cause of IRAP.[14,15,21,22] On the other hand, many studies including a study from our centre have shown a low prevalence of microlithiasis ranging from 6%–16%.[23–25] In a study of 75 patients with IRAP, we found that only 10 (13%) patients had microlithiasis, 35 (47%) patients developed CP during the follow-up—10 (13%) patients had miscellaneous aetiologies and no cause was found in the remaining 18 (24%) patients. Ten of these 35 patients who developed CP had biliary microlithiasis—8 of these 10 patients had undergone cholecystectomy/endoscopic sphincterotomy, yet continued to have recurrent pancreatitis and developed CP.[25]
 This varied prevalence reflects the uncertainty regarding the causal relationship between microlithiasis and pancreatitis. Microliths could also be a consequence of pancreatitis. Oedema of the head of pancreas could lead to biliary compression and gallbladder hypomotility. Gallbladder stasis may also result from fasting, parenteral nutrition and weight loss. Gallbladder contents may be modified, and bile may become more lithogenic. Two important studies[14,15] which found a high prevalence, i.e. 67% and 73% of microlithiasis or biliary sludge in patients with idiopathic AP had patients with a mean age >50 years and among them >50% were women. This was in contrast to our study where the mean age was 31.9 years and 80% were men.
Older patients especially women are more likely to have biliary lithiasis than young men. Furthermore, patients in our study had a mean symptom duration of 30.42 months and yet had not shown overt gallstones on USG. Had microlithiasis been the cause of RAP, they should have developed gallstones by the time they were evaluated.
Therefore, microlithiasis should be suspected as a cause of AP, when it is demonstrated on imaging ideally on EUS, and LFTs are abnormal at the time of pancreatitis.
Sphincter of Oddi dysfunction (SOD)
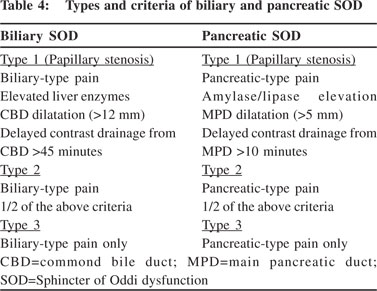
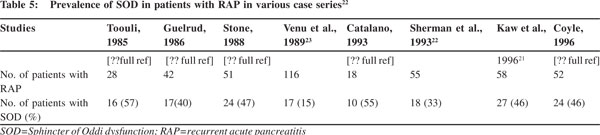
SOD is a benign obstructive disorder of the Sphincter of Oddi. It could be due to fibrosis, inflammation or hypertonic sphincter. SOD can be either biliary or pancreatic (Table 4). Pancreatitis in SOD usually results either from bile reflux into the pancreatic duct or pancreatic duct outflow obstruction.[23] SOD has been reported to be the cause of IRAP in up to one-third of cases but the results are quite variable (Table 5).[26,27] It has been reported predominantly from the USA and that too mainly from a few centres.
The diagnostic gold standard for SOD is Sphincter of Oddi manometry (SOM), which is performed endoscopically after cannulation of the papilla with a water perfused catheter or solid state catheter. In SOD, the basal sphincter pressure is >40 mmHg and high amplitude phasic contractions (>240 mmHg) occur more than 10/minute.28 Basal pressure usually predicts the response to sphincterotomy. However, satisfactory examination with SOM is achieved in only 50%–87% of patients. SOM is also associated with an incidence of 9%– 33% for post-procedure pancreatitis.[29]
This varied prevalence reflects the uncertainty regarding the causal relationship between microlithiasis and pancreatitis. Microliths could also be a consequence of pancreatitis. Oedema of the head of pancreas could lead to biliary compression and gallbladder hypomotility. Gallbladder stasis may also result from fasting, parenteral nutrition and weight loss. Gallbladder contents may be modified, and bile may become more lithogenic. Two important studies[14,15] which found a high prevalence, i.e. 67% and 73% of microlithiasis or biliary sludge in patients with idiopathic AP had patients with a mean age >50 years and among them >50% were women. This was in contrast to our study where the mean age was 31.9 years and 80% were men.
Older patients especially women are more likely to have biliary lithiasis than young men. Furthermore, patients in our study had a mean symptom duration of 30.42 months and yet had not shown overt gallstones on USG. Had microlithiasis been the cause of RAP, they should have developed gallstones by the time they were evaluated.
Therefore, microlithiasis should be suspected as a cause of AP, when it is demonstrated on imaging ideally on EUS, and LFTs are abnormal at the time of pancreatitis.
Sphincter of Oddi dysfunction (SOD)
SOD is a benign obstructive disorder of the Sphincter of Oddi. It could be due to fibrosis, inflammation or hypertonic sphincter. SOD can be either biliary or pancreatic (Table 4). Pancreatitis in SOD usually results either from bile reflux into the pancreatic duct or pancreatic duct outflow obstruction.[23] SOD has been reported to be the cause of IRAP in up to one-third of cases but the results are quite variable (Table 5).[26,27] It has been reported predominantly from the USA and that too mainly from a few centres.
The diagnostic gold standard for SOD is Sphincter of Oddi manometry (SOM), which is performed endoscopically after cannulation of the papilla with a water perfused catheter or solid state catheter. In SOD, the basal sphincter pressure is >40 mmHg and high amplitude phasic contractions (>240 mmHg) occur more than 10/minute.28 Basal pressure usually predicts the response to sphincterotomy. However, satisfactory examination with SOM is achieved in only 50%–87% of patients. SOM is also associated with an incidence of 9%– 33% for post-procedure pancreatitis.[29]
 SOD may involve either predominantly biliary sphincter or pancreatic sphincter or both. Pancreatic SOD is more likely to be associated with IRAP. SOD should be suspected in patients with IRAP only after other causes have been excluded.
SOD may involve either predominantly biliary sphincter or pancreatic sphincter or both. Pancreatic SOD is more likely to be associated with IRAP. SOD should be suspected in patients with IRAP only after other causes have been excluded.
Pancreas divisum (PD)
PD is the commonest congenital variant of the pancreas. It is reported to occur in 5%–14% of the general population including in India.[30] It occurs due to failure of the fusion of embryological ventral and dorsal ducts in utero. Therefore, most of the pancreas drains via the dorsal duct through the minor papilla. Usually, <5% patients become symptomatic. The symptomatic patients include those with stenosis of the minor papilla and those with santorinicoele, which is cystic dilatation of the distal dorsal duct just proximal to the minor papilla. Santorinicoele results from relative distal obstruction and weakness of the distal ductal wall. Although PD has been associated with IAP, there is a great deal of controversy on its role in the aetiopathogenesis of IAP. The arguments in favour of causal association of PD with IAP are: (i) the frequency of PD is much higher in patients with IRAP than the general population,[31] (ii) PD can cause AP due to relative obstruction to the flow of pancreatic juice from the dorsal duct through the minor papilla,[32] (iii) presence of pathological changes confined to the dorsal pancreas in patients with pancreatitis and PD, and (iv) therapeutic response to surgical/endoscopic sphincterotomy of the minor papilla by relieving the obstruction.[33,34] On the other hand, arguments against such a causal association are: (i) PD is asymptomatic in the vast majority of individuals,[35–37] (ii) there is no significant difference in the prevalence of PD between the pancreatitis and non-pancreatitis patient groups in a large series of patients undergoing ERCP,[38] and (iii) the response to minor papilla sphincterotomy is variable and suboptimal in the majority of patients presenting with RAP and CP.[39,40]
The prevalence of PD in RAP varies across various studies (Table 6).[25,35–37,41–43] A recent community-based study from Japan included 504 subjects from the general population, and 46 patients with pancreatitis (8 AP, 23 CP and 15 RAP). PD was present in 35% of patients with pancreatitis versus 2.6% in the general community and had an odds ratio of 23.4 for inducing pancreatitis.[44]
However, recent studies have shown an association of genetic mutations and PD in patients with IRAP. We have shown that SPINK1 (pancreatic secretory trypsin inhibitor) gene mutations were present in 40% of patients with IRAP and PD.[45] In addition, many CFTR (cystic fibrosis transmembrane regulator) gene polymorphisms were also found in them. These were similar to the mutations seen in patients with IRAP without PD and those with CP. Other studies have also shown that CFTR gene mutations are more frequently present in patients with IRAP and PD. These studies have suggested that PD alone is unlikely to be the cause of IRAP and it requires another hit, i.e. genetic mutations to cause pancreatitis. PD can be diagnosed on ERCP or magnetic resonance cholangiopancreatography (MRCP) where dorsal pancreatic duct is seen crossing the CBD anteriorly, and is separated from the small ventral duct. MRCP being as sensitive as ERCP is preferred for the diagnosis because of its non-invasive nature.[46]
Other congenital variants associated with IRAP Other congenital variants associated with RAP include anomalous pancreaticobiliary ductal union (APBDU), choledochocoele, choledochal cyst, duodenal duplication, and annular pancreas.
ABPDU is present in 1.5%–3% of individuals. There is anomalous union of the pancreatic and CBD outside the duodenal wall with a long common channel >15 mm. Because of this, there is reflux of bile into the MPD. AP can occur in 3%–31% of patients with ABPDU.[47]
 About 16% of type I and IV choledochal cysts may be associated with pancreatitis.[48] Type III choledochal cyst/ choledochocoele is dilatation of the intraduodenal segment of the CBD.[49] PD outflow may be obstructed by the dilated CBD or by the contents of the choledochocoele. Treatment involves deroofing of the choledochocoele by endoscopic sphincterotomy.
Annular pancreas: Annular pancreas is defined as a part of the pancreatic tissue partially or completely encircling the duodenum usually at the level of or just proximal to the major papilla.[50] It is often associated with duodenal or biliary obstructive symptoms and/or pancreatitis that may affect the annulus or the remaining pancreas. Treatment consists of gastrojejunostomy in case of duodenal occlusion. Meandering main pancreatic duct (MMPD): Meandering main pancreatic duct (MMPD) consists of either a loop-type or a reverse Z-type MPD. A recent study showed that the rate of MMPD was significantly higher in patients with idiopathic pancreatitis as compared to that in the community.[51]
Pancreaticobiliary tumours
Malignancy as a cause of AP should be suspected in any patient who presents with AP of unknown aetiology after 50 years of age. About 5%–7% patients with pancreaticobiliary tumours may present with AP.[52] Common tumours include ampullary tumours and cystic neoplasms of the pancreas, especially intraductal papillary mucinous tumour (IPMT). EUS has the highest sensitivity for identifying small pancreatic neoplasms.
About 16% of type I and IV choledochal cysts may be associated with pancreatitis.[48] Type III choledochal cyst/ choledochocoele is dilatation of the intraduodenal segment of the CBD.[49] PD outflow may be obstructed by the dilated CBD or by the contents of the choledochocoele. Treatment involves deroofing of the choledochocoele by endoscopic sphincterotomy.
Annular pancreas: Annular pancreas is defined as a part of the pancreatic tissue partially or completely encircling the duodenum usually at the level of or just proximal to the major papilla.[50] It is often associated with duodenal or biliary obstructive symptoms and/or pancreatitis that may affect the annulus or the remaining pancreas. Treatment consists of gastrojejunostomy in case of duodenal occlusion. Meandering main pancreatic duct (MMPD): Meandering main pancreatic duct (MMPD) consists of either a loop-type or a reverse Z-type MPD. A recent study showed that the rate of MMPD was significantly higher in patients with idiopathic pancreatitis as compared to that in the community.[51]
Pancreaticobiliary tumours
Malignancy as a cause of AP should be suspected in any patient who presents with AP of unknown aetiology after 50 years of age. About 5%–7% patients with pancreaticobiliary tumours may present with AP.[52] Common tumours include ampullary tumours and cystic neoplasms of the pancreas, especially intraductal papillary mucinous tumour (IPMT). EUS has the highest sensitivity for identifying small pancreatic neoplasms.
Metabolic factors
Hypertriglyceridaemia (HTG; >1000 mg/dL) and hypercalcaemia can cause recurrence of pancreatitis, if not identified and treated. HTG or chylomicronaemia may be responsible for 1%– 7% of all cases of pancreatitis.[53] Mutations in the lipoprotein lipase (LPL) gene have also been identified in patients with HTG-induced pancreatitis.[54] In HTG-related CP, Chang et al.[55] have shown a higher frequency of CFTR gene mutations suggesting that the mechanism of pancreatitis may be related to genetic predisposition. In their study of 126 patients with HTG, 13 (10.3%) carried a CFTR gene mutation. The CFTR gene mutation rate was significantly higher in those with pancreatitis than those without pancreatitis (26.1% [12 of 46] vs. 1.3% [1 of 80]; p <0.0001). Treatment primarily consists of lipid-lowering agents.
The prevalence of AP in hyperparathyroidim has been estimated to be between 1.5% and 13%. It can cause AP, RAP and CP. In a study of patients with primary hyperparathyroidism, 4 (16%) out of 25 patients with pancreatitis carried the N34S missense mutation in the SPINK1 gene, while all 50 controls (hyperparathyroidism without pancreatitis) showed no mutation in SPINK1 or PRSS1 genes (p<0.05 vs.controls, p<0.001 vs. general population).[56] In addition, CFTR gene mutations were present in 4 patients (p<0.05 vs. general population), while one patient carried a 5T allele. Treatment consists of parathyroidectomy.
Genetic risk factors
Many mutations have been found to be associated with CP. These include autosomal dominant mutations (N29I and R122H) of PRSS1 (cationic trypsinogen) gene, which is associated with hereditary pancreatitis with 80% penetrance.[57] Mutations in other genes, i.e. SPINK1 gene and CFTR are associated with idiopathic pancreatitis. [58,59]
Since patients with CP usually present with recurrent episodes of pain, they may not have evidence of chronicity in the initial course of illness and thus usually present with RAP in the beginning. Mutations in different genes thus may also be associated with IRAP. Indeed many studies have shown presence of mutations in these genes among patients with IRAP especially those with early onset of disease.[45,60,61]
Early chronic pancreatitis
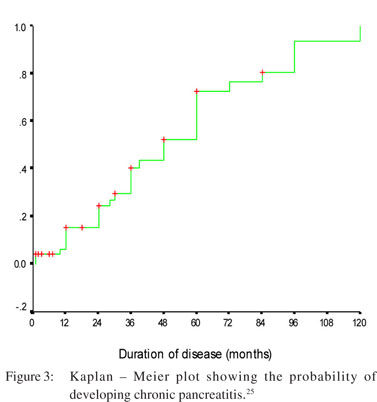
Early CP presents with recurrent attacks of AP. Initially, however, the features of chronicity are usually not evident on imaging, and become apparent only during further follow-up. It is quitelogical that patients destined to develop CP do so over a considerable period of time. During the period of evolution, patients experience recurrent episodes of acute inflammation which manifest clinically as RAP. The definite evidences of CP, i.e. calcification and/or ductal dilatation appear quite late. Thus, the patients are diagnosed to have RAP during this stage of evolution. The challenge is to recognize and diagnose early changes of CP at this stage because this will prevent many more unnecessary and expensive tests, and resolve the diagnostic dilemma. Recurrent episodes of inflammation leading to CP forms the basis of the “necrosis-fibrosis hypothesis”.[62] Clinically, though not every patient presents with RAP first and CP later. Many patients have features of CP during their first presentation and the reason for their silent behaviour during the period when they were developing inflammation and damage to the pancreas is not well understood. In previous studies it has been documented that 42%–45% of patients with RAP indeed have evidence of CP on imaging.[62] In a followup study of 75 patients with RAP at our centre, CP was documented in 47% of patients on long-term follow-up (Figure 3).[25]
Evaluation of a patient with RAP
Aetiological evaluation after the first episode of AP includes a detailed history of alcohol intake, smoking, medication associated with AP, trauma, viral illness, exposure to toxins, and a family history of AP. A history suggestive of cystic fibrosis in the form of chest symptoms, male infertility, etc. might provide useful information.
Investigative work-up: The investigations that are indicated for aetiology of AP may be divided into—phase I, phase II and phase III investigations.
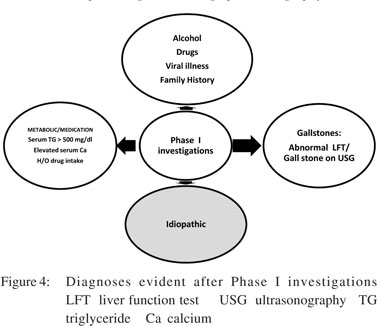
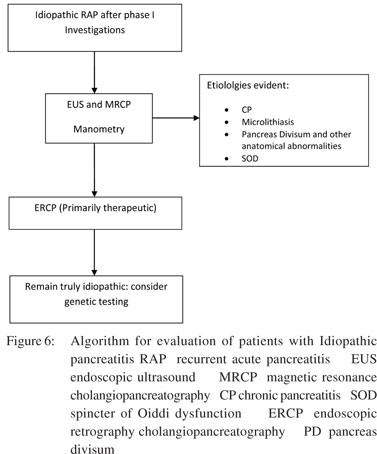
Phase I investigations: Phase I investigations should be carried out after the first episode of AP (Table 7). These include serum biochemistry and an abdominal USG. LFTs have a very important role during first 48 hours after onset of pancreatitis. Abnormal LFT may indicate biliary aetiology for AP. It has been shown that alamin aminotransferase (ALT) >3 times the upper limit of normal (ULN) and bilirubin level >3 mg/dL predict biliary pancreatitis with a high degree of accuracy. In a study, ALT level >150 IU/L had asensitivity of 96% for predicting biliary aetiology for AP.[63] One should strongly suspect microlithiasis as the cause of pancreatitis in presence of abnormal LFT and absence of gallstones on imaging. Serum calcium and triglyceride (TG) levels must be obtained after the subsidence of acute episode because they may not be elevated during the acute episode. If a patient have had a CECT scan of the abdomen during the acute episode, it need not be repeated at this stage. If the aetiology of pancreatitis is unclear after initial evaluation and phase I investigations, the patient is labelled as having idiopathic pancreatitis (Figure 4) and one should then proceed to phase II investigations (Table 7).

 Phase II investigations
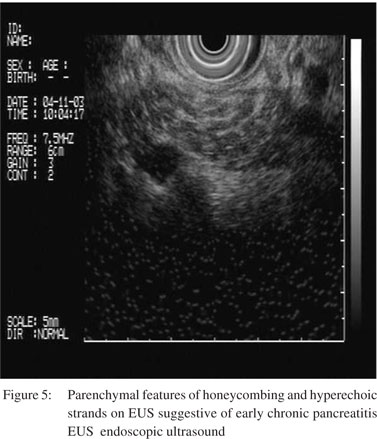
EUS: Over the years, EUS has replaced ERCP for diagnosing most of the aetiologies of idiopathic RAP including microlithiasis, occult biliary tumours, congenital anomaly of the pancreas, and CP. It has indeed become the imaging of choice for the evaluation of such patients with idiopathic pancreatitis. EUS has been documented to have a sensitivity of 96% for diagnosing microliths and has a negative predictive value of 95.4% for diagnosing CBD stones.[64] The most useful role of EUS is to diagnose early CP in patients who have presented with RAP and do not show evidence of chronicity on other imaging modalities such as computed tomography (CT) scan or MRCP. The changes suggestive of CP include both parenchymal (hyperechoic foci, hyperechoic strands, parenchymal lobularity, calcification) and ductal changes (pancreatic duct dilatation, pancreatic duct irregularity, hyperechoic pancreatic duct walls, visible pancreatic side branches) (Figure 5). Since EUS is quite sensitive to pick up subtle changes in the parenchyma, there is a possibility of over-diagnosis of CP by EUS. The sensitivity and specificity of EUS varies with the number of minimum criteria that is used as a cut-off for the diagnosis of CP. In a histological correlation study on surgically resected specimens in 71 patients, three or more EUS criteria provided the best balance of sensitivity (83.3%) and specificity (80%) for predicting pancreatic fibrosis.[65] The limitation of EUS is a lack of specificity, as some of the features of CP have been found in the elderly, men, persons with a history of alcohol abuse, smokers, and those with a history of AP.[66–71] A set of criteria for EUS diagnosis of CP have been laid down recently, to improve the diagnostic accuracy and reproducibility, based upon the consensus of 32 internationally recognized endosonographers, known as Rosemont criteria[72] Hyperechoic foci with shadowing, main pancreatic duct (MPD) calculi and lobularity with honeycombing have been defined as the major criteria, while the minor criteria for CP include cysts, dilated duct e”3.5 mm, irregular PD contour, dilated side branches e”1 mm , hyperechoic duct wall, strands, non-shadowing hyperechoic foci, and lobularity with non-contiguous lobules. On the basis of these criteria, the findings are classified as “consistent with CP”, “suggestive of CP”, “indeterminate for CP”, or “normal”. Accuracy of EUS can be improved by rational application of this valuable test, interpretation of EUS findings in the clinical context and results of other imaging findings. Increasing the minimum number of criteria required for the diagnosis can increase the accuracy in conditions where the suspicion of CP is low.
Phase II investigations
EUS: Over the years, EUS has replaced ERCP for diagnosing most of the aetiologies of idiopathic RAP including microlithiasis, occult biliary tumours, congenital anomaly of the pancreas, and CP. It has indeed become the imaging of choice for the evaluation of such patients with idiopathic pancreatitis. EUS has been documented to have a sensitivity of 96% for diagnosing microliths and has a negative predictive value of 95.4% for diagnosing CBD stones.[64] The most useful role of EUS is to diagnose early CP in patients who have presented with RAP and do not show evidence of chronicity on other imaging modalities such as computed tomography (CT) scan or MRCP. The changes suggestive of CP include both parenchymal (hyperechoic foci, hyperechoic strands, parenchymal lobularity, calcification) and ductal changes (pancreatic duct dilatation, pancreatic duct irregularity, hyperechoic pancreatic duct walls, visible pancreatic side branches) (Figure 5). Since EUS is quite sensitive to pick up subtle changes in the parenchyma, there is a possibility of over-diagnosis of CP by EUS. The sensitivity and specificity of EUS varies with the number of minimum criteria that is used as a cut-off for the diagnosis of CP. In a histological correlation study on surgically resected specimens in 71 patients, three or more EUS criteria provided the best balance of sensitivity (83.3%) and specificity (80%) for predicting pancreatic fibrosis.[65] The limitation of EUS is a lack of specificity, as some of the features of CP have been found in the elderly, men, persons with a history of alcohol abuse, smokers, and those with a history of AP.[66–71] A set of criteria for EUS diagnosis of CP have been laid down recently, to improve the diagnostic accuracy and reproducibility, based upon the consensus of 32 internationally recognized endosonographers, known as Rosemont criteria[72] Hyperechoic foci with shadowing, main pancreatic duct (MPD) calculi and lobularity with honeycombing have been defined as the major criteria, while the minor criteria for CP include cysts, dilated duct e”3.5 mm, irregular PD contour, dilated side branches e”1 mm , hyperechoic duct wall, strands, non-shadowing hyperechoic foci, and lobularity with non-contiguous lobules. On the basis of these criteria, the findings are classified as “consistent with CP”, “suggestive of CP”, “indeterminate for CP”, or “normal”. Accuracy of EUS can be improved by rational application of this valuable test, interpretation of EUS findings in the clinical context and results of other imaging findings. Increasing the minimum number of criteria required for the diagnosis can increase the accuracy in conditions where the suspicion of CP is low.
 In a study of 168 patients with idiopathic pancreatitis, EUS identified abnormality in 80% of patients, of whom 62% had gallstones, sludge or microlithiasis.[73] EUS correctly established the cause in 92% of patients compared to the final diagnosis at surgery. Another study, which looked at the diagnostic utility of ERCP with SOM, bile microscopy, and EUS in patients with idiopathic and RAP showed that, EUS was useful in diagnosing biliary diseases and tumours, and was able to identify 9 patients with CP, which ERCP could not identify.[74] Another study showed that the diagnostic yield of EUS in idiopathic pancreatitis was 68%.[75] The diagnoses made on EUS were CP, microlithiasis, PD, and pancreatic malignancy. In two other studies of 44 and 18 patients, the diagnostic yield of EUS was 41% and 78%, respectively in diagnosing biliary diseases among patients with idiopathic pancreatitis.[76,77] Repiso Ortega et al.[78] evaluated 49 patients with idiopathic AP prospectively with EUS and MRCP. EUS could detect aetiology in a higher proportion of patients than MRCP (51% vs. 20%; p=0.001). Cholelithiasis and biliary sludge (24%) were the most frequent EUS diagnoses, and PD (8%) was the most frequent MRCP diagnosis. The yield of EUS was lower in patients who had had a previous cholecystectomy.
With regard to the timing of EUS, an important study of 300 patients by Yusoff et al.[79] [??au: new reference inserted; pls check] showed that EUS could identify causes of the pancreatitis in one-third of the patients in whom routine evaluation was negative. They also showed that there was no difference in the yield whether EUS was performed after first or recurrent episodes of pancreatitis, if CP was excluded. Therefore, it is generally recommended that most patients with idiopathic AP should undergo an EUS after the first episode of pancreatitis. It should usually be done 6–8 weeks after the acute episode because by that time the changes of AP are likely to resolve.
ERCP: ERCP was the first-line investigation for the aetiological work-up of AP before EUS came in practice. ERCP still plays an important role in diagnosing PD, subtle changes of CP, CBD stones, and SOD. It has a diagnostic yield of 38%– 78% in patients with idiopathic pancreatitis. The major limitation of ERCP is the risk of post-ERCP pancreatitis, which varies from 5% to 10% but is much higher in patients with SOD. ERCP may prove to be both diagnostic and therapeutic but it is not preferred after the first attack because of its invasive nature and associated risk of post-ERCP pancreatitis. ERCP is generally reserved for therapeutic purposes in patients with treatable aetiologies such as microlithiasis, CBD stones, PD, SOD, choledochocoele, etc.
MRCP: MRCP is as good as ERCP for the assessment of biliary tree (both intra- and extra-hepatic) and pancreatic duct. With the advent of secretin MRCP, aetiologies such as SOD and PD which were earlier diagnosed only by ERCP, can well be diagnosed by MRCP, as shown by Mariani et al.[80] Other aetiologies which can be picked up by MRCP include occult biliary tumours, congenital anomaly of the pancreas, and bile duct stones. [81] Secretin MRCP is also a good modality to diagnose early CP.[82]
Phase III investigations: Genetic testing may be undertaken but its role outside of research setting is unclear because of a lack of therapeutic benefit. It might be indicated in patients with a family history of pancreatitis and those with subtle changes of CP. Sweat chloride test may be done. Serum autoantibodies are usually not indicated. Autoimmune pancreatitis is an altogether different disease both from the point of pathophysiology and clinical presentation. It is not a cause of IRAP and hence serum IgG4 should not be routinely advised to rule out autoimmune pancreatitis in patients with IRAP.
Management of patients with RAP: The episodes of AP should be treated in the standard fashion regardless of the aetiology. Recurrence is prevented by removing the cause of pancreatitis, e.g. by treating microlithiasis appropriately as shown in previous studies. Ros et al.[14] had shown significant decline in the recurrence (from 66%–75% to <1%) after cholecystectomy in patients with proven microlithiasis. However, in patients with high surgical risk, endoscopic sphincterotomy or ursodeoxycholic acid are other treatment options for microlithiasis.[83] Following certain reports of high prevalence of microlithiasis in patients with idiopathic AP, cholecystectomy is being recommended even without demonstrating microlithiasis.[84] Laparoscopic cholecystectomy, a seemingly “innocuous procedure” also seems to have contributed to such a recommendation. However, cholecystectomy including laparoscopic cholecystectomy is associated with certain risks including death. Thus, cholecystectomy may not be justified without unequivocal evidence of microlithiasis.
Treatment for SOD includes biliary and/or pancreatic sphincterotomy, which might lead to relief in >50% patients.[85,86] Prophylactic pancreatic stenting for 2 weeks after sphincterotomy has shown to reduce the incidence of post- ERCP pancreatitis.[87] Adding pancreatic sphincterotomy to biliary sphincterotomy does not lead to a better outcome as shown in a randomized control trial by Cote et al.88 They randomized 69 patients with IRAP and pancreatic SOD to either biliary sphincterotomy alone or both biliary and pancreatic sphincterotomy. The recurrence of pancreatitis was similar in both the groups (48.5% vs. 47.2%). Patients with pancreatic SOD were more likely to have recurrence than those with normal SOM.
Treatment for PD consists of endoscopic or surgical therapy. Endoscopic therapy consists of minor papilla sphincterotomy with or without stenting (Table 8).[33,89–95]
In a study of 168 patients with idiopathic pancreatitis, EUS identified abnormality in 80% of patients, of whom 62% had gallstones, sludge or microlithiasis.[73] EUS correctly established the cause in 92% of patients compared to the final diagnosis at surgery. Another study, which looked at the diagnostic utility of ERCP with SOM, bile microscopy, and EUS in patients with idiopathic and RAP showed that, EUS was useful in diagnosing biliary diseases and tumours, and was able to identify 9 patients with CP, which ERCP could not identify.[74] Another study showed that the diagnostic yield of EUS in idiopathic pancreatitis was 68%.[75] The diagnoses made on EUS were CP, microlithiasis, PD, and pancreatic malignancy. In two other studies of 44 and 18 patients, the diagnostic yield of EUS was 41% and 78%, respectively in diagnosing biliary diseases among patients with idiopathic pancreatitis.[76,77] Repiso Ortega et al.[78] evaluated 49 patients with idiopathic AP prospectively with EUS and MRCP. EUS could detect aetiology in a higher proportion of patients than MRCP (51% vs. 20%; p=0.001). Cholelithiasis and biliary sludge (24%) were the most frequent EUS diagnoses, and PD (8%) was the most frequent MRCP diagnosis. The yield of EUS was lower in patients who had had a previous cholecystectomy.
With regard to the timing of EUS, an important study of 300 patients by Yusoff et al.[79] [??au: new reference inserted; pls check] showed that EUS could identify causes of the pancreatitis in one-third of the patients in whom routine evaluation was negative. They also showed that there was no difference in the yield whether EUS was performed after first or recurrent episodes of pancreatitis, if CP was excluded. Therefore, it is generally recommended that most patients with idiopathic AP should undergo an EUS after the first episode of pancreatitis. It should usually be done 6–8 weeks after the acute episode because by that time the changes of AP are likely to resolve.
ERCP: ERCP was the first-line investigation for the aetiological work-up of AP before EUS came in practice. ERCP still plays an important role in diagnosing PD, subtle changes of CP, CBD stones, and SOD. It has a diagnostic yield of 38%– 78% in patients with idiopathic pancreatitis. The major limitation of ERCP is the risk of post-ERCP pancreatitis, which varies from 5% to 10% but is much higher in patients with SOD. ERCP may prove to be both diagnostic and therapeutic but it is not preferred after the first attack because of its invasive nature and associated risk of post-ERCP pancreatitis. ERCP is generally reserved for therapeutic purposes in patients with treatable aetiologies such as microlithiasis, CBD stones, PD, SOD, choledochocoele, etc.
MRCP: MRCP is as good as ERCP for the assessment of biliary tree (both intra- and extra-hepatic) and pancreatic duct. With the advent of secretin MRCP, aetiologies such as SOD and PD which were earlier diagnosed only by ERCP, can well be diagnosed by MRCP, as shown by Mariani et al.[80] Other aetiologies which can be picked up by MRCP include occult biliary tumours, congenital anomaly of the pancreas, and bile duct stones. [81] Secretin MRCP is also a good modality to diagnose early CP.[82]
Phase III investigations: Genetic testing may be undertaken but its role outside of research setting is unclear because of a lack of therapeutic benefit. It might be indicated in patients with a family history of pancreatitis and those with subtle changes of CP. Sweat chloride test may be done. Serum autoantibodies are usually not indicated. Autoimmune pancreatitis is an altogether different disease both from the point of pathophysiology and clinical presentation. It is not a cause of IRAP and hence serum IgG4 should not be routinely advised to rule out autoimmune pancreatitis in patients with IRAP.
Management of patients with RAP: The episodes of AP should be treated in the standard fashion regardless of the aetiology. Recurrence is prevented by removing the cause of pancreatitis, e.g. by treating microlithiasis appropriately as shown in previous studies. Ros et al.[14] had shown significant decline in the recurrence (from 66%–75% to <1%) after cholecystectomy in patients with proven microlithiasis. However, in patients with high surgical risk, endoscopic sphincterotomy or ursodeoxycholic acid are other treatment options for microlithiasis.[83] Following certain reports of high prevalence of microlithiasis in patients with idiopathic AP, cholecystectomy is being recommended even without demonstrating microlithiasis.[84] Laparoscopic cholecystectomy, a seemingly “innocuous procedure” also seems to have contributed to such a recommendation. However, cholecystectomy including laparoscopic cholecystectomy is associated with certain risks including death. Thus, cholecystectomy may not be justified without unequivocal evidence of microlithiasis.
Treatment for SOD includes biliary and/or pancreatic sphincterotomy, which might lead to relief in >50% patients.[85,86] Prophylactic pancreatic stenting for 2 weeks after sphincterotomy has shown to reduce the incidence of post- ERCP pancreatitis.[87] Adding pancreatic sphincterotomy to biliary sphincterotomy does not lead to a better outcome as shown in a randomized control trial by Cote et al.88 They randomized 69 patients with IRAP and pancreatic SOD to either biliary sphincterotomy alone or both biliary and pancreatic sphincterotomy. The recurrence of pancreatitis was similar in both the groups (48.5% vs. 47.2%). Patients with pancreatic SOD were more likely to have recurrence than those with normal SOM.
Treatment for PD consists of endoscopic or surgical therapy. Endoscopic therapy consists of minor papilla sphincterotomy with or without stenting (Table 8).[33,89–95]
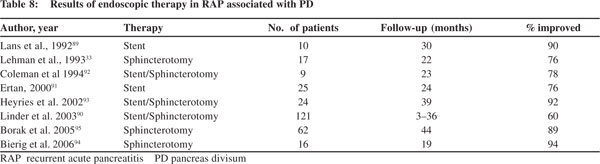 The efficacy of endoscopic therapy has varied from 50% to 90%.[94–98] Surgical sphincteroplasty has also shown an overall success rate of ~80%.96 There are no studies comparing surgical vs. endoscopic therapy for PD, and both of them seem to be equally effective but endoscopic sphincterotomy is preferred because it is minimally invasive. Both of them have shown better results in patients with RAP as compared to patients with CP or patients who only have pancreatic type of pain. Thereason why many patients with PD and IRAP may not respond well to minor papilla sphincterotomy is that genetic mutations might be more important in causing pancreatitis in such patients.[45,97]
There is a significant role of antioxidants in reducing relapses in patients with early CP presenting with recurrent pain.[101]
Summary
Approximately 20%–30% of patients with AP do not have a detectable cause after initial evaluation. These patients have a high risk of recurrence of pancreatitis. Patients with IRAP must be thoroughly evaluated to find out the aetiology. Microlithiasis is not a common cause of IRAP at least among Indian patients. The role of PD is better understood now and it is believed to be a cofactor; the main factor being associated genetic mutations. The role of SOD as a cause of IRAP remains controversial especially that of type II and type III and there is still not much clarity about the differential role of biliary and pancreatic SOD. Malignancy should be ruled out in any patient with idiopathic pancreatitis who is >50 years of age. Early CP can present initially as RAP. The work-up of patients with IRAP includes a detailed history and investigations. LFT, serum calcium, serum TG, abdominal USG and CECT scan are the standard phase I investigations. EUS, MRCP and possibly ERCP are indicated in phase II, if the work-up is negative after phase I tests. EUS should be done usually 6–8 weeks after an acute episode and is considered as an extremely useful modality. Phase III tests are not required often outside of research setting (Figure 6) [?au: pls check]. Genetic counselling may be appropriate in certain situations before genetic testing. The treatment of patients with IRAP is aimed at a specific aetiology. In general, empirical cholecystectomy should be discouraged with the availability and widespread use of EUS. Endoscopic sphincterotomy should be advised, if there is strong suspicion of SOD. Minor papilla sphincterotomy should be carried out in those with PD but with limited expectation as this may not be effective in a significant percentage of patients. Regular followup should be advised for patients with IRAP because most of them are likely to develop CP in due course of time which will require appropriate management.
The efficacy of endoscopic therapy has varied from 50% to 90%.[94–98] Surgical sphincteroplasty has also shown an overall success rate of ~80%.96 There are no studies comparing surgical vs. endoscopic therapy for PD, and both of them seem to be equally effective but endoscopic sphincterotomy is preferred because it is minimally invasive. Both of them have shown better results in patients with RAP as compared to patients with CP or patients who only have pancreatic type of pain. Thereason why many patients with PD and IRAP may not respond well to minor papilla sphincterotomy is that genetic mutations might be more important in causing pancreatitis in such patients.[45,97]
There is a significant role of antioxidants in reducing relapses in patients with early CP presenting with recurrent pain.[101]
Summary
Approximately 20%–30% of patients with AP do not have a detectable cause after initial evaluation. These patients have a high risk of recurrence of pancreatitis. Patients with IRAP must be thoroughly evaluated to find out the aetiology. Microlithiasis is not a common cause of IRAP at least among Indian patients. The role of PD is better understood now and it is believed to be a cofactor; the main factor being associated genetic mutations. The role of SOD as a cause of IRAP remains controversial especially that of type II and type III and there is still not much clarity about the differential role of biliary and pancreatic SOD. Malignancy should be ruled out in any patient with idiopathic pancreatitis who is >50 years of age. Early CP can present initially as RAP. The work-up of patients with IRAP includes a detailed history and investigations. LFT, serum calcium, serum TG, abdominal USG and CECT scan are the standard phase I investigations. EUS, MRCP and possibly ERCP are indicated in phase II, if the work-up is negative after phase I tests. EUS should be done usually 6–8 weeks after an acute episode and is considered as an extremely useful modality. Phase III tests are not required often outside of research setting (Figure 6) [?au: pls check]. Genetic counselling may be appropriate in certain situations before genetic testing. The treatment of patients with IRAP is aimed at a specific aetiology. In general, empirical cholecystectomy should be discouraged with the availability and widespread use of EUS. Endoscopic sphincterotomy should be advised, if there is strong suspicion of SOD. Minor papilla sphincterotomy should be carried out in those with PD but with limited expectation as this may not be effective in a significant percentage of patients. Regular followup should be advised for patients with IRAP because most of them are likely to develop CP in due course of time which will require appropriate management.
References
- Eland IA, Sturkenboom MJ, Wilson JH, Stricker BH. Incidence and mortality of acute pancreatitis between 1985 and 1995. Scand J Gastroenterol. 2000;35:1110–16.
- Spanier BW, Dijkgraaf MG, Bruno MJ. Epidemiology, aetiology and outcome of acute and chronic pancreatitis: an update. Best Pract Res Clin Gastroenterol. 2008;22:45–63.
- Shen HN, Lu CL, Li CY. Epidemiology of first-attack acute pancreatitis in Taiwan from 2000 through 2009: a nationwide population-based study. Pancreas. 2012;41:696–702.
- Lowenfels AB, Maisonneuve P, Sullivan T. The changing character of acute pancreatitis: epidenmiology, etiology, and prognosis. Curr Gastroenterol Rep. 2009;1:97–103.
- Peery AF, Dellon ES, Lund J, Crockett SD, McGowan CE, Bulsiewicz WJ, et al. Burden of gastrointestinal disease in the United States: 2012 update. Gastroenterology. 2012;143:1179–87.e1–3.
- Forsmark CE, Baillie J, AGA Institute Clinical Practice and Economics Committee; AGA Institute Governing Board. AGA Institute technical review on acute pancreatitis. Gastroenterology. 2007;132:2022–44.
- Garg PK, Madan K, Pande GK, Khanna S, Sathyanarayan G, Bohidar NP, et al. Association of extent and infection of pancreatic necrosis with organ failure and death in acute necrotizing pancreatitis. Clin Gastroenterol Hepatol. 2005;3:159–66.
- Isenmann R, Rau B, Beger HG. Bacterial infection and extent of necrosis are derterminants of organ failure in patients with acute necrotizing pancreatitis. Br J Surg. 1999;86:1020–4.
- Sharma, M, Banerjee D, Garg PK. Characterization of newer subgroups of fulminant and subfulminant pancreatitis associated with a high early mortality. Am J Gastroenterol. 2007;102:2688–95.
- Zhang W, Shan HC, Gu Y. Recurrent acute pancreatitis and its relative factors. World J Gastroenterol. 2005;11:3002–4.
- Gullo L, Migliori M, Pezzilli R, Oláh A, Farkas G, Levy P, et al. An update on recurrent acute pancreatitis: data from five European countries. Am J Gastroenterol. 2002;97:1959–62.
- Gao YJ, Li YQ, Wang Q, Li SL, Li GQ, Ma J, et al. Analysis of the clinical features of recurrent acute pancreatitis in China. J Gastroenterol. 2006;41:681–5.
- Garg PK, Goindi G, Tandon RK. Stimulation of gallbladder by intravenous infusion of amino acid: a new method to obtain duodenal bile for bile analyses. Dig Dis Sci. 2000;45:904–8.
- Ros E, Navarro S, Bru C, Garcia-Pugés A, Valderrama R. Occult microlithiasis in ‘idiopathic’ acute pancreatitis: prevention of relapses by cholecystectomy or ursodeoxycholic acid therapy. Gastroenterology. 1991;101:1701–9.
- Lee SP, Nicholls JF, Park HZ. Biliary sludge as a cause of acute pancreatitis. N Engl J Med. 1992;326:589–93.
- Neoptolemos JP, Davidson BR, Winder AF, Vallance D. Role of duodenal bile crystal analysis in the investigation of ‘idiopathic’ pancreatitis. Br J Surg. 1988;75:450–3.
- Buscail L, Escourrou J, Delvaux M, Guimbaud R, Nicolet T, Frexinos J, et al. Microscopic examination of bile directly collected during endoscopic cannulation of the papilla. Utility in patients with suspected microlithiasis. Dig Dis Sci 1992;37:116–20.
- Dahan P, Andant C, Levy P, Amouyal P, Amouyal G, Dumont M, et al. Prospective evaluation of endoscopic ultrasonography and microscopic examination of duodenal bile in the diagnosis of cholecystolithiasis in 45 patients with normal conventional ultrasonography. Gut. 1996;38:277–81.
- Opie EL. The etiology of acute hemorrhagic pancreatitis. Bull Johns Hopkins Hosp. 1901;12:182–8.
- Grau F, Almela P, Aparisi L, Bautista D, Pascual I, Pena A, et al. Usefulness of alanine and aspartate aminotransferases in the diagnosis of microlithiasis in idiopathic acute pancreatitis. Int J Pancreatol. 1999;25:107–11.
- Kaw M, Verma R, Brodmerkel GJ. ERCP, biliary analysis, sphincter of Oddi manometry (SOM) in idiopathic pancreatitis (IP) and response to endoscopic sphincterotomy (ES) [abstract]. Am J Gastroenterol. 1996;91:1935A.
- Sherman S, Jamidar P, Reber H. Idiopathic acute pancreatitis (IAP): endoscopic approach to diagnosis and therapy [abstract]. Am J Gastroenterol. 1993;88:1541A.
- Venu RP, Geenen JE, Hogan W, Stone J, Johnson GK, Soergel K. Idiopathic recurrent pancreatitis. An approach to diagnosis and treatment. Dig Dis Sci 1989;34:56–60.
- Nash JA, Geenen JE, Hogan WJ, Geenen DJ, Catalano MF, Johnson GK, et al. The role of sphincter of Oddi manometry (SOM) and biliary microscopy in evaluating idiopathic recurrent pancreatitis (IRP) [abstract]. Gastroenterology. 1996;110:31A.
- Garg PK, Tandon RK, Madan K. Is biliary microlithiasis a significant cause of idiopathic recurrent acute pancreatitis? A long-term follow-up study. Clin Gastroenterol Hepatol. 2007;5:75–9.
- Sherman S, Ruffolo TA, Hawes RH, Lehman GA. Complications of endoscopic sphincterotomy. A prospective study with emphasis on the increased risk associated with sphincter of Oddi dysfunction and nondilated bile ducts. Gastroenterology. 1991;101:1068–75.
- Clain JE, Pearson RK. Evidence-based approach to idiopathic pancreatitis. Curr Gastroenterol Rep. 2002;4:128–34.
- Hogan WJ, Geenen JE, Dodds WJ. Dysmotility disturbances of the biliary tract: classification, diagnosis, and treatment. Semin Liver Dis 1987;7:302–10.
- Tarnasky P, Cunningham J, Cotton P, Hoffman B, Palesch Y, Freeman J, et al. Pancreatic sphincter hypertension increases the risk of post-ERCP pancreatitis. Endoscopy. 1997;29:252–7.
- Sahni D, Jit I, Harjeet. Gross anatomy of the pancreatic ducts in north Indians. Trop Gastroenterol. 2001;22:197–201.
- Gregg JA. Pancreas divisum: its association with pancreatitis. Am J Surg. 1977;134:539–43.
- Sahel J, Cros RC, Bourry J, Sarles H. Clinico-pathological conditions associated with pancreas divisum. Digestion. 1982;23:1–8.
- Lehman GA, Sherman S, Nisi R, Hawes RH. Pancreas divisum: results of minor papilla sphincterotomy. Gastrointest Endosc. 1993;39:1–8.
- Delhaye M, Engelholm L, Cremer M. Pancreas divisum: controversial clinical signiûcance. Dig Dis. 1988;6:30–9.
- Cotton PB. Congenital anomaly of pancreas divisum as cause of obstructive pain and pancreatitis. Gut. 1980;21:105–14.
- Richter JM, Schapiro RH, Mulley AG, Warshaw AL. Association of pancreas divisum and pancreatitis, and its treatment by sphincteroplasty of the accessory ampulla. Gastroenterology. 1981;81:1104–10.
- Bernard JP, Sahel J, Giovannini M, Sarles H. Pancreas divisum is a probable cause of acute pancreatitis: a report of 137 cases. Pancreas. 1990;5:248–54.
- Harig JM, Hogan WJ. Pancreas divisum: a case against surgical treatment. Adv Surg. 1988;21:111–26.
- Russell RC, Wong NW, Cotton PB. Accessory sphincterotomy (endoscopic and surgical) in patients with pancreas divisum. Br J Surg. 1984;71:954–7.
- Whitcomb DC, Gorry MC, Preston RA, Furey W, Sossenheimer MJ, Ulrich CD, et al. Hereditary pancreatitis is caused by a mutation in the cationic trypsionogen gene. Nat Genet. 1996;14:141–5.
- Brenner P, Duncombe V, Ham JM. Pancreatitis and pancreas divisum: aetiological and surgical considerations. Aust N Z J Surg. 1990;60:899–903.
- Morgan DE, Logan K, Baron TH, Koehler RE, Smith JK. Pancreas divisum: implications for diagnostic and therapeutic pancreatography. AJR Am J Roentgenol. 1999;173:193–8.
- Fischer M, Fogel EL, McHenry L, Sherman S, Watkins JL, Schmidt S, et al. ERCP/manometry in 1,108 idiopathic pancreatitis patients. Gastrointest Endosc. 2005;61:190A.
- Gonoi W, Akai H, Hagiwara K, Akahane M, Hayashi N, Maeda E, et al. Pancreas divisum as a predisposing factor for chronic and recurrent idiopathic pancreatitis: initial in vivo survey. Gut. 2011;60:1103–8.
- Garg PK, Khajuria R, Kabra M, Shastri SS. Association of SPINK1 gene mutation and CFTR gene polymorphisms in patients with pancreas divisum presenting with idiopathic pancreatitis. J Clin Gastroenterol. 2009;43:848–52.
- Matos C, Metens T, Deviere J, Delhaye M, Le Moine O, Cremer M, et al. Pancreas divisum: evaluation with secretin-enhanced magnetic resonance cholangiopancreatography. Gastrointest Endosc. 2001;53:728–33.
- Sugiyama M, Atomi Y, Kuroda A. Pancreatic disorders associated with anomalous pancreaticobiliary junction. Surgery. 1999;126:492–7.
- Visser BC, Suh I, Way LW, Kang SM. Congenital choledochal cysts in adults. Arch Surg. 2004;139:855–60; discussion 860–2.
- Ladas SD, Katsogridakis I, Tassios P, Tastemiroglou T, Vrachliotis T, Raptis SA. Choledochocele, an overlooked diagnosis: report of 15 cases and review of 56 published reports from 1984 to 1992. Endoscopy. 1995;27:233–9.
- Urayama S, Kozarek R, Ball T, Brandabur J, Traverso L, Ryan J, et al. Presentation and treatment of annular pancreas in an adult population. Am J Gastroenterol. 1995;90:995–9.
- Gonoi W, Akai H, Hagiwara K, Akahane M, Hayashi N, Maeda E, et al. Meandering main pancreatic duct as a relevant factor to the onset of idiopathic recurrent acute pancreatitis. PLoS One. 2012;7:e37652.
- Kahrilas PJ, Hogan WJ, Geenen JE, Stewart ET, Dodds WJ, Arndorfer RC. Chronic recurrent pancreatitis secondary to a submucosal ampullary tumor in a patient with neurofibromatosis. Dig Dis Sci. 1987;32:102–7.
- Fortson MR, Freedman SN, Webster PD 3rd. Clinical assessment of hyperlipidemic pancreatitis. Am J Gastroenterol. 1995;90:2134–9.
- Jap TS, Jenq SF, Wu YC, Chiu CY, Cheng HM. Mutations in the lipoprotein lipase gene as a cause of hypertriglyceridemia and pancreatitis in Taiwan. Pancreas. 2003;27:122–6.
- Chang YT, Chang MC, Su TC, Liang PC, Su YN, Kuo CH, et al. Association of cystic fibrosis transmembrane conductance regulator (CFTR) mutation/variant/haplotype and tumor necrosis factor (TNF) promoter polymorphism in hyperlipidemic pancreatitis. Clin Chem. 2008;54:131–8.
- Felderbauer P, Karakas E, Fendrich V, Bulut K, Horn T, Lebert R, et al. Pancreatitis risk in primary hyperparathyroidism: relation to mutations in the SPINK1 trypsin inhibitor (N34S) and the cystic fibrosis gene. Am J Gastroenterol. 2008;103:368–74.
- Ulrich CD 2nd, Kopras E, Wu Y, Ward S. Hereditary pancreatitis: epidemiology, molecules, mutations, and models. J Lab Clin Med. 2000;136:260–74.
- Midha S, Khajuria R, Shastri S, Kabra M, Garg PK. Idiopathic chronic pancreatitis in India: phenotypic characterisation and strong genetic susceptibility due to SPINK1 and CFTR gene mutations. Gut. 2010;59:800–7.
- Garg PK. Chronic pancreatitis in India and Asia. Curr Gastroenterol Rep. 2012;14:118–24.
- Masamune A, Ariga H, Kume K, Kakuta Y, Satoh K, Satoh A, et al. Genetic background is different between sentinel and recurrent acute pancreatitis. J Gastroenterol Hepatol. 2011;26:974–8.
- Sultan M, Werlin S, Venkatasubramani N. Genetic prevalence and characteristics in children with recurrent pancreatitis. J Pediatr Gastroenterol Nutr. 2012;54:645–50.
- Comfort MW, Gambill EE, Baggenstoss AH. Chronic relapsing pancreatitis: a study of 29 cases without associated disease of the biliary or gastrointestinal tract. Gastroenterology. 1946;6:376–408.
- Tenner S, Dubner H, Steinberg W. Predicting gallstone pancreatitis with laboratory parameters: a meta-analysis. Am J Gastroenterol. 1994;89:1863–6.
- Amouyal P, Palazzo L, Amouyal G, Pansot P, Mompoint D, Vilgrain V, et al. Endosonography: Promising method for diagnosis of extrahepatic cholestasis. Lancet. 1989;2:1195–8.
- Varadarajulu S, Eltoum I, Tamhane A, Eloubeidi MA. Histopathologic correlates of noncalcific chronic pancreatitis by EUS: a prospective tissue characterization study. Gastrointest Endosc. 2007;66:501–9.
- Wiersema MJ, Hawes RH, Lehman GA, Kochman ML, Sherman S, Kopecky KK. Prospective evaluation of endoscopic ultrasonography and endoscopic retrograde cholangiopancreatography in patients with chronic abdominal pain of suspected pancreatic origin. Endoscopy. 1993;25:555–64.
- Bhutani MS. Endoscopic ultrasonography: changes of chronic pancreatitis in asymptomatic and symptomatic alcoholic patients. J Ultrasound Med. 1999;18:455–62.
- Yusoff IF, Sahai AV. A prospective, quantitative assessment of the effect of ethanol and other variables on the endosonographic appearance of the pancreas. Clin Gastroenterol Hepatol. 2004;2:405–9.
- Rajan E, Clain JE, Levy MJ, Norton ID, Wang KK, Wiersema MJ, et al. Age-related changes in the pancreas identified by EUS: a prospective evaluation. Gastrointest Endosc. 2005;61:401–6.
- Sahai AV, Zimmerman M, Aabakken L, Tarnasky PR, Cunningham JT, van Velse A, et al. Prospective assessment of the ability of endoscopic ultrasound to diagnose, exclude, or establish the severity of chronic pancreatitis found by endoscopic retrograde cholangiopancreatography. Gastrointest Endosc. 1998;48:18–25.
- Wallace MB, Hawes RH, Durkalski V, Chak A, Mallery S, Catalano MF, et al. The reliability of EUS for the diagnosis of chronic pancreatitis: interobserver agreement among experienced endosonographers. Gastrointest Endosc. 2001;53:294–9.
- Catalano MF, Sahai A, Levy M, Romagnuolo J, Wiersema M, Brugge M, et al. EUS-based criteria for the diagnosis of chronic pancreatitis: the Rosemont classification. Gastrointest Endosc 2009;69:1251–61.
- Frossard JL, Sosa-Valencia L, Amouyal G, Marty O, Hadengue A, Amouyal P. Usefulness of endoscopic ultrasonography in patients with “idiopathic” acute pancreatitis. Am J Med. 2000;109:196–200.
- Coyle WJ, Pineau BC, Tarnasky PR, Knapple WL, Aabakken L, Hoffman BJ, et al. Evaluation of unexplained acute and acute recurrent pancreatitis using endoscopic retrograde cholangiopancreatography, sphincter of Oddi manometry and endoscopic ultrasound. Endoscopy. 2002;34:617–23.
- Tandon M, Topazian M. Endoscopic ultrasound in idiopathic acute pancreatitis. Am J Gastroenterol. 2001;96:705–9.
- Norton SA, Alderson D. Endoscopic ultrasonography in the evaluation of idiopathic acute pancreatitis. Br J Surg. 2000;87:1650–5.
- Liu CL, Lo CM, Chan JK, Poon RT, Fan ST. EUS for detection of occult cholelithiasis in patients with idiopathic pancreatitis. Gastrointest Endosc. 2000;51:28–32.
- Repiso Ortega A, Gómez-Rodríguez R, Romero M, Fernandez- Zapardiel S, Del Mar Cespedes M, Carrobles JM. Prospective comparison of endoscopic ultrasonography and magnetic resonance cholangiopancreatography in the etiological diagnosis of “idiopathic” acute pancreatitis. Pancreas. 2011;40:290–4.
- Yusuff IF, Raymod G, Sahai AV. A Prospective comparison of the yield of EUS in primary vs. Recurrent idiopathic acute pancreatitis. Gastrointest Endosc. 2004;60:673–8.
- Mariani A, Curioni S, Zanello A, Passaretti S, Masci E, Rossi M, et al. Secretin MRCP and endoscopic pancreatic manometry in the evaluation of sphincter of Oddi function: a comparative pilot study in patients with idiopathic recurrent pancreatitis. Gastrointest Endosc. 2003;58:847–52.
- Taourel P, Bret PM, Reinhold C, Barkun AN, Atri M. Anatomic variants of the biliary tree: diagnosis with MR cholangiopancreatography. Radiology. 1996;199:521–7.
- Czakó L. Diagnosis of early-stage chronic pancreatitis by secretin—enhanced magnetic resonance cholangiopancreatography. J Gastroenterol. 2007;42:113–7.
- Steinberg W, Tenner S. Acute pancreatitis. N Engl J Med. 1994;330:1198–210.
- Draganov P, Forsmark CE. “Idiopathic” pancreatitis. Gastroenterology. 2005;128:756–63.
- Geenen JE, Hogan WJ, Dodds WJ, Toouli J, Venu RP. The efficacy of endoscopic sphincterotomy after cholecystectomy in patients with sphincter-of-Oddi dysfunction. N Engl J Med. 1989;320:82–7.
- Raddawi HM, Geenen JE, Hogan WJ, Dodds WJ, Venu RP, Johnson GK. Pressure measurements from biliary and pancreatic segments of sphincter of Oddi. Comparison between patients with functional abdominal pain, biliary, or pancreatic disease. Dig Dis Sci. 1991;36:71–4
- Tarnasky PR, Palesch YY, Cunningham JT, Mauldin PD, Cotton PB, Hawes RH. Pancreatic stenting prevents pancreatitis after biliary sphincterotomy in patients with sphincter of Oddi dysfunction. Gastroenterology. 1998;115:1518–24.
- Cote GA, Imperiale TF, Schmidt SE, Fogel E, Lehman G, McHenry L, et al. Similar effects of biliary, with or without pancreatic, sphincterotomy in treatment of idiopathic recurrent acute pancreatitis. Gastroenterology. 2012;143:1502–9.
- Lans JI, Geenen JE, Johanson JF, Hogan WJ. Endoscopic therapy in patients with pancreas divisum and acute pancreatitis: a prospective, randomized, controlled clinical trial. Gastrointest Endosc. 1992;38:430–4.
- Linder JD, Bukeirat FA, Geenen JE, Catalano MF; American Society for Gastrointestinal Endoscopy. Long-term response to pancreatic duct stent placement in symptomatic patients with pancreas divisum. Gastrointest Endosc. 2003;57:208A.
- Ertan A. Long-term results after endoscopic pancreatic stent placement without pancreatic papillotomy in acute recurrent pancreatitis due to pancreas divisum. Gastrointest Endosc. 2000;52:9–14.
- Coleman SD, Eisen GM, Troughton AB, Cotton PB. Endoscopic treatment in pancreas divisum. Am J Gastroenterol. 1994;89:1152–5.
- Heyries L, Barthet M, Delvasto C, Zamora C, Bernard JP, SahelJ. Long-term results of endoscopic management of pancreas divisum with recurrent acute pancreatitis. Gastrointest Endosc. 2002;55:376–81.
- Bierig L, Chen YK, Shah RJ. Patient outcomes following minor papilla endotherapy (MPE) for pancreas divisum (PD). Gastrointest Endosc. 2006;63:313A.
- Borak G, Alsolaimon M, Holt E, Romagnuolo J, Hawes RH, Cotton PB. Pancreas divisum: long-term follow up after endoscopic therapy. Gastrointest Endosc. 2005;61:149A.
- Keith RG, Shapero TF, Saibil FG, Moore TL. Dorsal duct sphincterotomy is effective long-term treatment of acute pancreatitis associated with pancreas divisum. Surgery. 1989;106:660–6; discussion 666–7.
- Bertin C, Pelletier AL, Vullierme MP, Bienvenu T, Rebours V, Hentic O, et al. Pancreas divisum is not a cause of pancreatitis by itself but acts as a partner of genetic mutations. Am J Gastroenterol. 2012;107:311–17.
- Bhardwaj P, Garg PK, Maulik SK, Saraya A, Tandon RK, Acharya SK. A randomized controlled trial of antioxidant supplementation for pain relief in patients with chronic pancreatitis. Gastroenterology. 2009;136:149–59.
|
|
|
|
|
|