48uep6bbphidvals|391
48uep6bbph|2000F98CTab_Articles|Fulltext
Introduction
Caroli’s disease was first described by Jacques Caroli in as a rare disease with saccular dilatation of intrahepatic ducts that causes bile stasis and intrahepatic duct stones.[1] The disorder can involve all intrahepatic ducts or one part of the liver.[2] The mode of inheritance is still unknown but in the majority of cases it is transmitted in an autosomal recessive fashion.[3] One recent observation in a family from Japan suggests an autosomal dominant mode of inheritance.[4]
Case Report
A 2 years and 7 months old female Sudanese child, presented with right upper quadrant pain and vomiting since 8 days, after which she developed dark discoloration of urine, pale stools, anorexia and lethargy followed by yellowish discoloration of the body. She was the only child of non-consanguineous parents. She has no previous medical or surgical illness, and her parents were apparently healthy. On physical examination she was afebrile, icteric, had no palpable lymph nodes, weighed 11.8 kg, and had a height of 95 cm which werel at 10th and 50th percentile, respectively. Her abdomen was initially soft, lax, and non-tender with diminished bowel sounds. Her liver was palpable 6 cm below the right costal margin in the right midclavicular line, with a span of 7 cm. It was firm and had a smooth surface with well-defined margins. She had no splenomegaly, both kidneys were not palpable and no other mass was felt. There was no ascites. Later she developed abdominal tenderness on deep palpation.
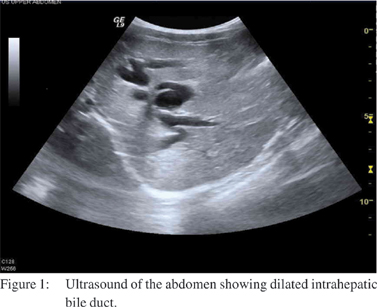
Laboratory investigation showed Hb of 12gm/L, WBC count of 8.3x103/mm3, platelet count of 406x103/mm3 and ESR of 2mm/ hr, urea, creatinine, Na, K, PT, PTT and albumin were normal. Initially her amylase was high 777 U/L, after 5 days it decreased to 582 then to 42. Total bilirubin was 0.58mg/dL with direct bilirubin being 0.33 mg/dL; ALT (alanine aminotransferase) was elevated at 166 u/L. Triglycerides were 50 mg/dL, and cholesterol 216 mg/dL. Hepatitis B surface antigen was negative, and so were anti-hepatitis C, and anti-mumps IgM titers. Chest X-ray showed unremarkable cardiac size and lung fields, plain abdominal X-ray showed non-specific bowel gas distribution with no fluid level, her abdominal ultrasound showed cystic dilatation of the common bile duct (CBD) with dimensions of 4.6x1.7 cm with intrahepatic biliary radical dilatation. There was no evidence of gall stones. Kidneys and spleen were unremarkable and no other masses were seen as shown in Figure 1.
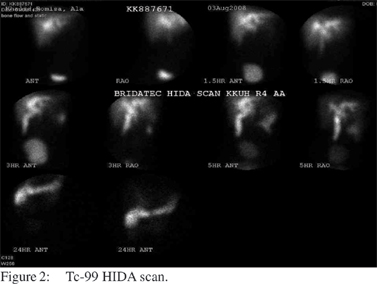
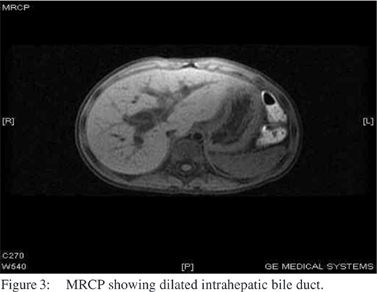
Tc-99m HIDA scan showed adequate hepatocyte function, dilated biliary channels and a dilated gall bladder. Slow hepatobiliary transit of tracer was also noted (Figure 2). The patient underwent endoscopic ultrasonography which showed normal pancreas, normal pancreatic duct, a shrunken gall bladder with thickened wall, cystic dilatation of CBD, dilated hepatic duct, minimal intrahepatic duct dilatation and minimal sludge in the CBD. MRCP showed a diffuse fusiform dilatation of the CBD and intrahepatic biliary ducts. No definite distal obstructive lesions were noted. No underlying liver lesions or CBD stones were found. The pancreas was unremarkable; and overall findings were suggestive of Caroli’s disease (Figure 3).
With the clinical examination, laboratory findings and radiological imaging, a diagnosis of Caroli’s disease with acute pancreatitis was entertained in this child. The child was managed conservatively and was discharged with follow-up in the clinic.
Discussion
Caroli’s disease occurs in two form (i) pure form called Caroli’s disease characterized by ectasia of intrahepatic bile ducts without other histologic abnormalities and (ii) combined formcalled Caroli syndrome in which ectasia of intrahepatic bile duct is associated with congenital hepatic fibrosis and renal cystic disease.[5] Both conditions result from malformation of embryonic ductal plate at different levels of the biliary tree.[6] Although duct abnormalities are assumed to be present at birth, patients are often asymptomatic until the teenage years or later.[4]
The disorder is slightly more predominant among females as seen in our patient.[6] The most common symptoms are fever, abdominal pain, jaundice and dark urine (caused by cholangitis or gall bladder stones) and hepatomegaly. Most of these symptoms were seen in our patient, however she had no splenomegaly, but she presented with pancreatitis. In some cases it could start as portal hypertension with splenomegaly and variceal bleeding (caused by congenital hepatic and fibrosis).[7] Patients may present with acute pancreatitis but is very rare.[2]
There are some reports of its association with other abnormalities such as autosomal recessive polycystic kidney disease,[8] duodenal diverticulitis, testicular ectopia,hypospadia, the Laurence-Moon-Biedl syndrome,[7] and the oralfacial- digital syndrome type 1 associated with cystic renal disease and Caroli’s disease has also been reported.[9] Caroli’s disease is complicated by septicemia, cholangitis, gall bladder stones, biliary abscess and cholangio-carcinoma.[6] None of these association conditions or complications were noted on our patient.
Blood test typically shows an elevated level of alkaline phosphate and gamma-glutamyl transferase. Ultrasonography and computed tomography are helpful in showing diffuse or focal intrahepatic bile duct dilatation.[10] CT scan shows central dot sign in these patients which is suggested as pathognomic finding in Caroli’s disease. The fibrovascular bundles containing portal vein radical and a branch of hepatic artery bridging the saccule, appear as central dots or linear streak as seen in our patient.[5] Transhepatic cholangiogaphy is used for diagnostic confirmation, MRCP is a non-invasive modality that has proven itself as the best diagnostic method because of its ability to show communications between cyst ducts and biliary tree[10] but it has difficulties in the differentiation of cholangiocarcinoma, Caroli’s disease and secondary sclerosing cholangitis.[11] MRCP shows diffuse fusiform dilation of the CBD and intrahepatic bile ducts as seen in our patient. Liver biopsy in Caroli’s disease shows secondary biliary cirrhosis and dilated bile duct containing inspissated bile and a thickened wall caused by marked chronic inflammation, while preserving normal lobular architecture.[12,13] Histopathology shows intrahepatic bile duct ectasia and proliferation, associated with severe peripheral fibrosis and confirms the congenital hepatic fibrosis component of Caroli’s syndrome. Liver biopsy was not done in our patient. The treatment is supportive; cholangitis is treated with appropriate antibiotics. Gall bladder stones are treated by litholytic therapy or therapeutic endoscopic retrograde cholangiography (ERC).[13] Most patients are treated with ursodeoxycholic acid in an attempt to reduce the formation of intrahepatic stones which are rich in cholesterol. In localized disease, good results have been reported with partial hepatectomy,[14] and liver transplantation is the most effective current treatment for diffuse forms. Orthotropic liver transplantation (OLT) is indicated in decompensated liver disease, bilobar disease, recurrent episodes of cholangitis and secondary biliary cirrhosis. OLT is a curative therapy in this group of patients.[15,16] Patients who underwent combined liver / kidney transplantation had a better outcome of patient survival (100%) at 5 years than those who underwent liver transplantation alone with patient survival at 5 years being 75.4%.[17]
Poor prognosis in recipients associated with congenital hepatic fibrosis appears to be related to the presence of perioperative cholangitis.15 Prognosis is fairly good, unless recurrent cholangitis and renal failure develops. Tsuchida et al reviewed 50 well documented cases, and at least 46% of these patients had poor prognosis and died as a result of sepsis, hepatic abscesses, hepatic failure or portal hypertension.[4]
Acknowledgements
The authors wish to thank Ms. Loida M. Sese for secretarial assistance.
References
1. Agustsson AI, Cariglia N. [Caroli’s disease, case report and review of the literature]. Laeknabladid. 2007;93:603–5.
2. Pezzilli R, Carini G, Cennamo V. Education and imaging. Hepatobiliary and pancreatic: Caroli’s disease. J Gastroenterol Hepatol. 2008;23:1621.
3. Behrman RE, Kliegman RM, Jenson HB, editors. Nelson Textbook of Paediatrics. 17th ed. Philadelphia: Saunders; 2004.p.1343.
4. Tsuchida Y, Sato T, Sanjo K, Etoh T, Hata K, Terawaki K, et al. Evaluation of long-term results of Caroli’s disease: 21 years’ observation of a family with autosomal “dominant” inheritance, and review of the literature. Hepatogastroenterology. 1995;42:175–81.
5. Gupta AK, Gupta A, Bhardwaj VK, Chansoria M. Caroli’s disease. Indian J Pediatr. 2006;73:233–5.
6. Suchy FJ, Sokol RJ, Balistreri WF. Liver disease in children. 3rd ed. New York: Cambridge University Press; 2007.p.933.
7. de Tommaso AM, Santos DS, Hessel G. Caroli’s disease: 6 case studies. Acta Gastroenterol Latinoam. 2003;33:47–51.
8. Kim JT, Hur YJ, Park JM, Kim MJ, Park YN, Lee JS. Caroli’s syndrome with autosomal recessive polycystic kidney disease in a two month old infant. Yonsei Med J. 2006;47:131–4.
9. Toprak O, Uzum A, Cirit M, Esi E, Inci A, Ersoy R, et al. Oralfacial- digital syndrome type 1, Caroli’s disease and cystic renal disease. Nephrol Dial Transplant. 2006;21:1705–9.
10. Aguilera V, Rayon M, Perez-Aguilar F, Berenguer J. Caroli’s syndrome and imaging: report of a case. Rev Esp Enferm Dig. 2004;96:74–6.
11. Weber C, Kuhlencordt R, Grotelueschen R, Wedegaertner U, Ang TL, Adam G, et al. Magnetic resonance cholangiopancreatography in the diagnosis of primary sclerosing cholangitis. Endoscopy. 2008;40:739–45.
12. Sylvestre PB, El-Youssef M, Freese DK, Ishitani MB. Caroli’s syndrome in a 1-year-old child. Liver Transpl. 2001;7:59.
13. Yonem O, Bayraktar Y. Clinical characteristics of Caroli’s syndrome. World J Gastroenterol. 2007;13:1934–7.
14. Karim AS. Caroli’s disease. Indian Pediatr. 2004;41:848–50.
15. Habib S, Shakil O, Couto OF, Demetris AJ, Fung JJ, Marcos A, et al. Caroli’s disease and orthotopic liver transplantation. Liver Transpl. 2006;12:416–21.
16. Wang ZX, Yan LN, Li B, Zeng Y, Wen TF, Wang WT. Orthotopic liver transplantation for patients with Caroli’s disease. Hepatobiliary Pancreat Dis Int. 2008;7:97–100.
17. Millwala F, Segev DL, Thuluvath PJ. Caroli’s disease and outcomes after liver transplantation. Liver Transpl. 2008;14:11–7.